HAL Id: hal-01611017
https://hal.archives-ouvertes.fr/hal-01611017
Submitted on 5 Sep 2018
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
Inverse gas chromatography a tool to follow
physicochemical modifications of pharmaceutical solids:
Crystal habit and particles size surface effects
María Graciela Cares Pacheco, Rachel Calvet, G. Vaca-Medina, A. Rouilly,
Fabienne Espitalier
To cite this version:
María Graciela Cares Pacheco, Rachel Calvet, G. Vaca-Medina, A. Rouilly, Fabienne Espitalier.
In-verse gas chromatography a tool to follow physicochemical modifications of pharmaceutical solids:
Crystal habit and particles size surface effects. International Journal of Pharmaceutics, Elsevier,
2015, 494 (1), pp.113-126. �10.1016/j.ijpharm.2015.07.078�. �hal-01611017�
Inverse
gas
chromatography
a
tool
to
follow
physicochemical
modifications
of
pharmaceutical
solids:
Crystal
habit
and
particles
size
surface
effects
M.G.
Cares-Pacheco
a,*
,
R.
Calvet
a,
G.
Vaca-Medina
b,c,
A.
Rouilly
b,c,
F.
Espitalier
aaUniversitédeToulouse;MinesAlbi,UMRCNRS5302,CentreRAPSODEE;CampusJarlard,F-81013Albicedex09,France bUniversitédeToulouse;INP-ENSIACET,LCA,310130Toulouse,France
cINRA;UMR1010CAI,310130Toulouse,France
Keywords:
D-mannitol
Polymorphism Surfaceenergy
Inversegaschromatography Spraydrying
Cryomilling ABSTRACT
Powders are complex systems and so pharmaceutical solids are not the exception. Nowadays, pharmaceuticalingredientsmustcomplywithwell-defineddraconianspecificationsimposingnarrow particlesizerange,controlonthemeanparticlesize,crystallinestructure,crystalhabitsaspectand surface properties of powders, among others. The different facets,physical forms, defectsand/or impuritiesofthesolidwillalteritsinteractionproperties.Apowerfulwayofstudyingsurfaceproperties isbasedontheadsorptionofanorganicorwatervaporonapowder.Inversegaschromatography(IGC) appearsasausefulmethodtocharacterizethesurfacepropertiesofdividedsolids.
TheaimofthisworkistostudythesensitivityofIGC,inHenry’sdomain,inordertodetecttheimpactof sizeandmorphologyinsurfaceenergyoftwocrystallineformsofanexcipient,D-mannitol.Surface
energyanalysesusingIGChaveshownthattheaformisthemostenergeticallyactiveform.Tostudysize andshapeinfluenceonpolymorphism,pureaandbmannitolsampleswerecryomilled(CM)and/or spraydried(SD).Allformsshowedanincreaseofthesurfaceenergyaftertreatment,withahigher influenceforbsamples(gd
sof40–62mJm!2)thanforamannitolsamples(gdsof75–86mJm!2).Surface
heterogeneityanalysisinHenry’sdomainshowedamoreheterogeneousb-CMsample(62–52mJm!2).
Moreover,despiteitssphericalshapeandquitehomogeneoussizedistribution,b-SDmannitolsamples showedaslightlyheterogeneoussurface(57–52mJm!2)also higherthantherecrystallizedb pure
sample("40mJm!2).
1.Introduction
Pharmaceuticalsolids mustcomply withwell-defined speci-ficationsintermsofbioavailability,solubility,toxicityandstability. Nowadays, the requirements are more and more draconian, imposingnarrowparticlesizerange,controlonthemeanparticle size, crystalline structure, crystal habits aspect and surface propertiesof powders,among others. Alargeset ofoperations is developed to answer these requirements. The processes for producingfinepowders(aroundmicrometer)arevariedasmelt quenching,grinding, freeze-drying,spray drying,crystallization, antisolventprecipitation,milling,andsupercriticalfluids. Depend-ing upon the nature of the active pharmaceutical ingredients (APIs),it is known that thepreparation methodinfluencesthe
physicalstabilityandcrystallizationbehavior.Theimpactofthese processes on solid phase transformations may lead to the formationofametastableoranamorphousform,oramixtures ofvariouscrystallineformsincludingotherhydrates(orsolvates). Thesechangesaredesiredforcertainstagesoftheformulationor forsomeusepropertiesoftheAPI,butsometimescanalsohave undesirableeffectsonthesolid.
Inversegaschromatography(IGC)appearsasatooltostudythe changes on surface properties in order to highlight process influencestoassessdrugdeliverysystemsperformance. Mechani-cal operations are the most studied processes to determine pharmaceuticalsolidssurface’sbehavior.Mostauthorsdescribed an increase of the dispersive component of the solid surface energy,
g
ds,aftermechanical grinding(Table1).Thisincreaseis
generallyattributedtotheexposureofspecificcrystalfacetsthat presentdifferentchemicalgroups,theformationofhigherenergy zonessuchascrystaldefects,dislocationsand/ortosolidsstate transformations(Chamarthyet Pinal, 2008; Feeley etal., 2002;
* Correspondingauthor.
Fengetal.,2008;Hengetal.,2006;Hoetal.,2012;Newelland Buckton,2004).
1.1.Surfaceenergy,particlessizeandsurfacechemistry
Duetotheanisotropicnatureofpowders,theexhibitionofnew crystalfacesundermillingcanchangetheacidic/basiccharacterof the solid depending on the functional groups present in the exposed facets. Heng et al. (2006) highlighted the anisotropic natureofformIparacetamolcrystals.Theconfrontationofsessile dropandIGC,allowedthemtoconcludethatgrindingleadstoa fragmentation of thefacet(010),which possesses theweakest attachmentenergyandexhibitsthelarge
g
ds.Thus,aftermillingthe
g
dsofthesamplesincreased,showingamorehydrophobicsurface,
withdecreasingparticlesize.
York et al. (1998) were interested in the milling of DL
-propranololhydrochloride.Theevolutionof
g
dsshowedtodepend
on the particles size. During milling the surface becomes increasinglymoreenergetic(gd
sfrom45to61mJ/m2forparticles
75–16.5
mm)
untilreachedaplateaufollowedbyasmallfalling
dsforthefinestpowder(<10
mm).
Theauthorsconcludedthatthe increasesing
ds,andin
Dg
adsusingCH2Cl2asprobemolecule,are duetoafragmentationreleasingthedominantcrystalface,which possesthelowestattachmentenergyandisrichinnaphthalene groups. Attrition might become significantas millingintensity increase releasingfaceshavingOHgroups,naphthaleneand Cl! ions.Trowbridgeetal.(1998)highlightedthatacetaminophensize reductionfrom30to10
mm
leadedtoanincreaseofg
dsfrom50.9to
61.3mJ/m2andtoanincreaseof
Dg
adsfrom327to506J/mol,using chloroformasprobemolecule.Millingalsoincreasesthe hydro-phobicandbasiccharacterofacetaminophensurface.Thisresults
are in agreement with those obtained by molecular modeling whichestablishedthatmillingleadstotheexposureofthecrystal facet (010) which contain an hydrophobic methyl group, a benzeneringandacarboxylgroup,bothbasic.
OhtaandBuckton(2004)studiedsurfaceenergeticchangesof cefditorenpivoxil,acephalosporinantibiotic,asconsequenceof milling.After grinding in a vibration mill,the authorsfound a decreaseinthe
g
dsaccordingtothegrindingtime,from52.3mJ/m2before milling to 45.8mJ/m2 after 30min of grinding with a decreaseinsolidcrystallinity.Inaddition,theauthorshaveshown adecreaseinthesolidacidiccharacter withanincreasedonits basicity.Theseeffects areattributedtotheexposureofcarbonyl groups,whichhaveanelectrondonatingnature.
Luneretal.(2012)studiedbyIGCtheimpactofhighshearwet milling(HSWM)anddrymilling(DM)onthesurfacepropertiesof two pharmaceutical compounds, succinic acid and sucrose. Physicochemical characterizationof both samples showed that bulk properties were unaffectedby wet and dry milling while surfacepropertiesanalysesshowedanincreaseofsolidsdispersive surface energyafter DMand HSWM.Succinic acidsamples,
g
d s=35mJ/m2,exhibitminordifferencesbetweendrymilledandwet milled samples, 40#2mJ/m2, attributed to minimal impact of cleavageandtheexposure ofcrystal facetswithsimilaratomic surface arrangements. For HSWM sucrose, the polarity of the solvents used during wet milling influenced
g
ds of the milled
samplesfrom55to71to91mJ/m2,for hexane,methyltertiary butyl ether and ethanol respectivelywhile dry milled samples exhibita
g
dsof44mJ/m2.Differencesbetweendryandwetmilling
processeswereattributedtotheattritionmechanisminpresence ofsolvent.
ReducingparticlesizemayalsobenecessaryfortheAPItoreach thetargetorgan,particularlywhenthedrugadministrationisby Nomenclature
as(m2/g) Specificsurfaceareaofthesolid d(n,0.5)(mm) Numbermediandiameter D[y,0.5](mm) Medianvolumediameter
Dg
ads(J/mol) Molarfreeenergyvariation foran isother-maladsorptionofprobemolecules m(g) Samplemassn(mol) Desorbedmolenumber
nads(mmol/g) Adsorbedmolenumberpergramofsolid nm(mol) Monolayercapacityornumberofadsorbed
molescorrespondingtoamonolayer P(Pa) Vaporpressureorpartialpressure Psat(Pa) Saturationvaporpressure T(K) Temperature
Tc(K) Columntemperature tN(min) Netretentiontime VN(cm3) Netretentionvolume
Wadh(J/m2) Workofadhesionwhenadsorptionoccurs n/nm(–) Surfacecoverage
Greeksymbols
g
dl (J/m2orN/m) Liquidsurfaceenergy(orsurfacetension)
g
ds(J/m2orN/m) Dispersive component of solid surface
energy
g
s(J/m2orN/m) Totalsurfaceenergyofasolidg
sps (J/m2orN/m) Specific component of solid surfaceenergy
u
s(–) SurfacecoverageTable1
Anoverviewoftheinfluenceofparticlessizereductionoverthesurfaceproperties ofpharmaceuticalingredientsbyIGC.
Pharmaceuticalsolid gd
s Reference
Acetaminophen " Trowbridgeetal.(1998)
Hengetal.(2006)
Cefditorenpivoxil # OhtaandBuckton(2004)
DL-propanololhydrochloride " Yorketal.(1998)
Felodipine " ChamarthyandPinal(2008)
Griseofulvine
ChamarthyandPinal(2008)
" Feng etal.(2008) OtteandCarvajal(2011)
# Otteet al.(2012)
Ibipinabant " Gambleetal.(2012)
Indomethacin " Planinseketal.(2010)
Limetal.(2013)
Lactose
Ahfatetal.(2000) Feeleyetal.(2002) NewellandBuckton(2004)
" Thielmannetal.(2007) Shariareetal.(2011) BrumandBurnett(2011) Jonesetal.(2012) Mannitol " Hoetal.(2012) Salbutamolsulfate " Ticehurstetal.(1994) Feeleyetal.(1998)
SalmeterolXinofoate " Tongetal.(2001,2006)
Dasetal.(2009)
Sucrose " SuranaHasegawaetal.et(2003)al.(2009) Luneretal.(2012)
inhalation. Nowadays, dry powder inhalers (DPIs) are of great interestthankstotheabsenceofpropellantandthestabilityofthe formulationasaresultofthedrystate.Asuccessfuldrugdelivery willdependontheinteractionbetweenthepowderformulation andthedeviceperformance.Duringinhalation,theAPIdetaching from the carrier by the energy of the inspired airflow that overcomestheadhesionforcesbetweentheAPIand thecarrier. Over the past few years, lactose has been considered as the excipientofchoiceinseveralsolidoraldosageformsandsomany studieshasbeencarriedonlactose,mostofthembasedonthe study of its physical–chemical properties as function of the manufacturingprocess(Pilceretal.,2012).Thus, whylactoseis alsothemoststudiedorganicsolidbyIGC.Feeleyetal.(2002)and
Shariareetal.(2011)studiedtheinfluenceofmillingonthesurface propertiesoflactose.IGCanalysesshowedalowsensitivityof
g
dsto
milling. However, the study of basic and amphoteric probes showed changes of the hydroxyl groups presented in lactose surface.Shariareetal.(2011)alsohighlightedthattheacidicor basiccharacterofthepowderseemstoberelatedtothesizeof startingparticles.Forsample,themicronizationoflargerparticles, 50–100
mm,
resultsinan increaseof thespecificcomponentof solidsurfaceenergy,g
sps,measuredwithTHF(basicprobe).Whilethemicronizationoffinerparticles,<20
mm,
leadstoanincreaseofg
sps usingamphotericprobesmoleculessuchasacetone.1.2.Surfaceenergyandcrystallinesolidstate
Tongetal. (2001, 2006)studied theinfluenceof solid–solid interactionsontheaerosol performanceofsalmeterolxinafoate (SX)polymorphsandlactosecarrierbyIGC.SXisahighlyselective bronchodilator, known to exist in two crystallineforms. Three activebatchesofSXweregenerated:thetwopolymorphs,SXIand SXII,crystallizedusingsolutionenhanceddispersionby supercrit-icalfluidsfrommethanolsolution(SEDS)andaformcalledMSXI generated by the micronisation of the SXI form. First of all, dispersivesurfaceenergyanalysisbyIGCexhibitsa moreactive MSXIsample(gd
SXI"33mJ/m2,gdSXII"29mJ/m2and
g
dMSXI"39mJ/m2).IGCwasalsoappliedtocalculatethecohesionbetweenSX samplesandtheadhesionbetweenthesamplesofSXandlactose. Thestudyof thestrengthdrug–drugcohesion anddrug-carrier adhesion suggests that the active particles of SX bond more stronglytothecarrierparticlesoflactosethattothoseoftheirown species, except for SXII–lactose: SXI–SXI (190.7MPa), SXII–SXII (67.3MPa),MSX–MSX(245.0MPa),SXI–lactose(212.6MPa)SXII– lactose(47.5MPa),MSXI–lactose(278.1MPa).Theuseoflactoseas acarrierimprovesaerosolperformanceabout25%forthebatchSXI and140%forthebatchMSXI.
Traini et al. (2008) studied by IGC the lactose–salbutamol sulfateinteractionsinDPIformulations.Theaimwastoinvestigate lactose pseudo-polymorphs,
a-anhydrous,
a-monohydrate
andb-anhydrous,
in terms of carrier functionality. In this work,a-monohydrate
formexhibitsbestaerosolperformance, indicat-ingthatcarriersurfacechemistryplaysadominatingroleinDPIs. The authors also highlighted an inverse relationship between surfaceenergyandaerosolefficiency.Morerecentlystudieshasbeencarriedoutinordertoprovidea substitutefor
a-lactose
monohydrateascarrierin DPI formula-tions.Indeed,lactosepossessesseveraldrawbacksduetoitsbovine origin,its incompatibilitywithamino groups likepeptides and proteinsanditstendencytobecomeamorphousaftermechanical treatment. Nowadays, mannitol appears to be an adequate substituteforlactosebecauseit doesnotcarry reducinggroups thatmaycausechemicalinteractionwithproteinsandishighly crystallineevenuponspraydrying(Maasetal.,2011).SurfaceenergyanalysisofD-mannitolhasbeenfocusedonthe
stable
b
form.IGChashighlighteditsacidicnature,attributedto thehighdensityofhydroxylgroupsatmannitolsurface(Saxena etal.,2007).Morerecentlyresearchwerefocusedontheeffectof millingand/orsurfaceenergyheterogeneityofthestableb
form (Hoetal.,2010, 2012).Our previousresearchwork,Caresetal. (2014),was focusedonthestudyofD-mannitolpolymorphs.D-mannitol pure polymorphs exhibit a more active and highly heterogeneous
a
form(74.9to45.5mJ/m2)withalessactiveand quitehomogeneousstableb
mannitol("40–38mJ/m2),whichalso behave similarly to the instabled
form. Mannitol particles, generated by SD, have been used commercially in bronchial provocationtest(Aridol).Tangetal.(2009)studiedtheimpactof different generation processes in particles to improve aerosol performance.Threepowdersampleswereprepared:byconfined liquidimpingingjets(CLIJs)followedbyjetmilling(JM),SDandJM. These processes generated quite different powder samples, needle-shape for CLIJs samples, spherical for SD samples and orthorhombique for theJM samples. Dispersivesurface energy analysisbyIGCexhibitsamoreactiveCLIJssampleattributedtothe presence ofa
mannitol (gdCLIJs"85mJ/m2,
g
dSD"60mJ/m2 andg
dJM"48mJ/m2). Particles shape showed to be an important
contributor totheaerosol performanceofmannitol powdersas itaffectsthesurfaceenergyandparticlesdynamics.
Yamauchi et al. (2011) studied physicochemical differences with emphasis on the surface properties, by IGC and DVS, of niclosamideandnaproxensodiumandtheirrespectiveanhydrate anddehydratedhydratedforms.Thenaproxensodiumanhydrate formshowedahigher
g
dsandhigherrateofmoisturesorptionthan
thedehydratedhydratedform,whereastheoppositewasobserved forniclosamide.
1.3.Surfaceenergy,defectsandamorphization
Many APIs are poorly soluble in water, which limits their bioavailability.Theirrateofdissolutionmayimprovebyusingan amorphous phase of the API. Indomethacin (IDMC) is a good example.IDMCisanantipyreticandanti-inflammatorydrugused inmanypharmaceuticalformulations,ithasfourpolymorphsand anamorphous.Accordingtothegrindingtemperature,belowor higher than its glass transition temperature (Tg), and grinding intensitythemoststableformatambientconditions,
g-IDMC,
can transformtothemetastablea
formorleadtoanamorphousstate (Crowleyand Zografi, 2002; Desprez and Descamps, 2006). As IDMCishighlyinsolubleinwater(0.02mg/mL),thebioavailability oftheproductanditsabsorptionbythegastrointestinaltractcan beimprovedbyusinganamorphousformofthedrug(Imaizumi etal.,1980).Planinseketal.(2010)studiedthecrystallinefraction thattransformsintoamorphousformsuponintensivemilling.IGC wasusedtodetectsurfacechangescausedbymillingcrystallineg-IDMC
whileDSCwasusedtodeterminethemass(orequivalent volume) fraction of the samples transformed. The authors determinedthatthedispersiveenergyofthestableg-IDMC
was 32.2mJ/m2andthatoftheamorphous,generatedinaballmillfor 120min,of43.3mJ/m2.Studiesontheinfluenceofmillingtime, allows theauthors toillustrate thesensitivity of theIGC-ID to detectcrystalsurfacedefectsashigh-energysites(gd30min=
g
d60min=g
damorphe).Bycomparingtimeevolutionoftheamorphousfraction
ofthesurfacebyIGCandvolumebyDSCtheauthorsshowedthat surfacetransformsathigherrate(anorderofmagnitude)thanthe bulkphase.IGCseemstobeapowerfultooltoquantifyamorphous rateandtolocateitbytheconfrontationwithbulk(orvolume) measurementtechniques(BrumandBurnett,2011).
Thedevelopmentofcost-efficienttechnologiesforthe genera-tionofdrugpowderswithdesiredphysicochemicalpropertiesis stillachallengeforpharmaceuticalcompanies.Toactuallydefinea successfulprotocolitseemsnecessarytostudysurfaceenergyasa functionofpolymorphism,surfacechemistryoftheexposedfacets, particlessizeandshape(crystalhabits).Nevertheless,ascanbe depicted, it is quite difficultto actually distinguish each effect influence.Theobjectiveofthisworkistoquantifytheeffectofsize andcrystalhabitsinsurfaceenergyofanAPI,D-mannitol.
Tostudyparticlessizeandshapeinfluenceonsurfaceenergetics ofD-mannitolpolymorphstwotechniquesareused:spraydrying
and/orcryomilling.Asmannitolisknowntobehighlycrystalline evenduringmillingorspraydrying,sizereductionprocessesusedin thisworkwereestablishedinwaysthatpolymorphssolidssamples donotundergoanyphasetransformation.IGCatinfinitedilution (IGC-ID)isusedto studysolidsurfaceanisotropyatlowsurface coverage,whileDVSwillbeusedtohaveamoreglobalviewofthe interactionpotential.Thedualaimofthisworkistohighlightthe impact of sizeand crystal habitsontheadsorption behavior of differentanhydrousformsofD-mannitolandtocomparethesurface
energydeterminationtechniquesusedpointingouttheirrelevance consideringthephysicalsenseofthemeasure.
2. Materialsandmethods 2.1.Materials
D-mannitolwasgenerouslyprovidedbyRoquette(France).The
batch, Pearlitol 160C, is composed of 99% of
b
mannitol mass percentageandonly1%ofsorbitol.Highpuritydeionizedwater (18MVcm)wasobtainedfromalaboratorypurificationsystem. Thenon-polarprobesoctane,nonaneanddecanewerepurchased fromSigma,assay>99%.2.2.Generationandformulationprotocols 2.2.1.Crystallization
Pure
b
anda
formsweregeneratedbyantisolventprecipitation (acetone)andbyseedingandfastcoolingrespectivelyasdescribed previouslyinCaresetal.(2014).2.2.2.Spraydrying(SD)
Theexperimentswereperformedusingalaboratoryminispray dryer BüchiB-290 equipped with a two-fluid nozzleof 0.5mm diameter.Theflowrateoftheaqueousmannitolsolutions,10g/L,is setat6mL/min andtheatomizingairrateat600NL/h.Airinlet temperatureissetto120$Cwhileoutlettemperaturewasmeasured
byatemperaturesensorintheairexitpoint,noted87$C.Torecover
the powder the aspiration is set to 35m3/h (maximum level). Sampleswerestoredatroomtemperatureundervacuuminglass desiccatorscontainingsilicagel(relativehumidityof6%).
2.2.3.Cryomilling(CM)
ARetschcryogenicimpactmillwasusedformilling.Samplesof around500mgwereplacedinstainlesssteelgrindingjarsof5mL containingonestainlesssteelgrindingballof5mmdiameter.The samples were milledfor 4 cycles in total, each consistingof a 10mingrindingtimeat25Hzfollowedbyintervalsof0.5minat 5Hz. Thesampleswere continuallycooled withliquidnitrogen beforeandduringthegrindingprocess.
2.3.Particlesphysicochemicalcharacterization 2.3.1.Chemicalpurity
ThechemicalpurityofeachsamplewascheckedbyHPLCusing a Hi-Plex Caligandexchange column coupledwitha refractive
index detector (analysis conditions: mobile phase HPLC grade water,flowrate0.3mLmin!1,columntemperature45$C).
2.3.2.Polymorphsfingerprints
Themannitolpowdersampleswereanalyzedusingdifferent techniquestoconfirmtheidentityofthedifferentpolymorphs. X-ray powder diffraction patterns (XRPD) were obtained using a PANanalytical X’Pert Pro MPD diffractometer (set-up Bragg– Brentano).Diffractiondataisacquiredbyexposingthepowders samplestoCu-Karadiationatavoltageof45kVandtoacurrentof 40mA.Thedatawerecollectedoverarangeof8–50$2u&atstep
size of 0.03$. Quality diffraction patterns were obtained in a
relativelyshortexposuretime(20min).Dataanalysisisdonewith theX’Pert Data Collector softwarewhile phase identificationis madewithPANalyticalHighScoreMoresoftwareanddatabases “ICDD Powder Diffraction File 2” and “Crystallography Open Database”.
FT-Raman spectra were recorded with an atomic force microscopeequipped witha confocal Ramanimaging upgrade, Alpha300fromWITec.Amagnificationof50%andanexcitation source of Nd:YAG (532nm) were used. The Raman data were acquired using WITec Project Plus software that hasa 16 bits resolutionandasamplingrateof250Hz.
Thermalanalysis was performed witha DSC Q200 fromTA Instrument.Samplesamountofabout3–5mgareplacedin non-hermeticaluminumpanels,inthetemperaturerangeof20–200$C
ataheatingrateof5$C/minundernitrogenatmosphere.Nitrogen
flow was adjusted at 50mL/min. All data measurements are averagesof atleast3 measureson3differentsamples.Heatof fusionvalues weredeterminedusinga sigmoidbaselinewitha standarddeviationoflessthan0.5%(6measuresaverage). 2.3.3.Sizeandparticleshapeanalysis
Particle shape analyses were examined using a XL30 SEM, scanning electron microscopy, witha field emission gun (FEG) operatingat15or20kV.Sampleswereplacedontodoubled-sided adhesiveandcoatedwithPlatinumfor4minusinganautosputter coaterSC7640underargongaspurge.
The particle size analysis was performed by image analysis usinganopticalbancPharmaVisionSystemPVS830fromMalvern Instruments.Toseparatetheparticles,apressureof6barwasset (SPD1300). Thenumber of particlestobe analyzedwas setat 70,000.Two objectifswereused“zoom3” whichcananalyzea particlesizerangebetween310and7.5
mm,
witharesolutionof 7mm
and “zoom 5” which allows to analyze a particle range between100and1.7mm
witharesolutionof3mm.
2.3.4.Surfaceanalysistechniques
SpecificsurfaceareaandsurfaceenergyanalysesofD-mannitol
polymorphs were carried out using an IGC–Surface Energy Analyser(IGC–SEA)fromSMS.Eachpowdersamplewaspacked intopre-silanisedcolumnsof300mmlengthand4mminternal diameter, plugged with silanised glass wool. Prior to analysis, eachcolumnwasconditionedat50$Cfor 2hat0%RH.Helium
wasusedasacarriergasat10sccmandthecolumntemperature was fixed at 30$C during the analysis, all experiences were
undertakeninidenticalconditions.Theretentiontimesofprobe moleculesandmethaneweredeterminedusing aflame ioniza-tion detector (FID).Deadvolume wasdeterminedby methane injections and dispersive surface energy determination by a homologous series of n-alkanes (n-decane, n-nonane and n-octane).Dispersivesurfaceenergyprofileswerecalculatedusing Cirrus Plus software (version 1.2.3.2 SMS, London) following Dorris-Grayapproach.Thecorrespondingsurfacecoverage,
u
s,at each injection concentrationis theratiobetween thedesorbedamount,n,determinedfrompeakareaandthemonolayercapacity ofthesolid,nm(us=n/nm).Inourexperiments,nmwasdetermined fromthespecificsurfaceareavalueobtainedbyIGC–SEAusing n-nonane as probe molecule. Full details of the experimental procedure and analysis by IGC have beenpublishedpreviously (Caresetal.,2014).
3. Results
3.1.D-mannitoldepartsamples
3.1.1.Powdercrystallinityandpurity
XPRDpatterns of thecommercial formP160C and the pure forms
b
anda
mannitolareshowninFig.1.Asitisdepictedfrom thefigure,a
mannitol hascharacteristic reflectionpeaksat 2u positionsof13.7$, 17.3$and19.9$and21.4$whileforb
mannitolarelocated at 14.7$, 18.8$, 23.6$ and 29.5$. No differences in the
characteristicreflectionpeaksbetweenthecommercialpowder
Pearlitol160Cand the
b
pureformwerefound, thedifferences betweenpatternspeaksintensitycanbeattributedtomorphology differencesbetweenbothsamples.Indeed,sorbitolpresencesin the commercialpowder P.160C is not detected byXPRD but is detectedbyDSC(Table2).AsshowninFig.2,thefirstendothermic peakat81.4$C,isconsistentwiththemeltingpointofa-sorbitol
(Nezzal et al., 2009) which decreasesboth heat of fusion and meltingpointof
b
polymorph.It should be noted that the detection of small quantities is complexandmaydependonthelocationofsorbitolwithinthe powder,neartothesurfaceorwithinthebulkofthecrystals. 3.1.2.Particlessizeandhabit
As described in our previous work, the commercialsample Pearlitol 160C (P160C) is composed by irregular sticks with a medianvolume diameterD[y,0.5]of 64
mm
and a D[y,0.9]of 250mm
(Fig.3a).Theb
formgeneratedbyantisolvent precipita-tion is formedby better-defined sticks withsmoother surfaces (Fig.3b), and it has a D[y, 0.5]of 26.7mm
and a D[y, 0.9]of 81.7mm.
Itseemsthatforbothsamples,thesmallerparticlesget stuckonthesurfaceofthelargestones.The
a
formisthefinestonewithneedle-shapemorphologyand aD[y,0.5]of25.3mm
andaD[y,0.9]of42.4mm
(Fig.3c).Itshould benoticethatparticlessizeanalysisusingPVS830seemstobenot welladaptedforthea
formanalysis.InfactSEManalysisshoweda narrowersizedistribution.ItispossiblethatPVS830characterizes particlesagglomerationsdue tohighercohesionforcesbetween theseparticles. 10 15 20 25 30 35 40 0 1 2 3 4 5 6 7 8 x 104 2θ Intensity P.160C (1) β (2) α (3) (1) (2) (3)Fig. 1.X-raypatternsofdepartsamples,aandbmannitolandthecommercialform Pearlitol160C.
Table2
Summaryofphysicalsolid-statecharacterizationobtainedbyDSC.
P160C b-form a-form
Heatoffusion(Jg!1) 300.2 304.5 294.4
0.4 – –
Meltingpoint(onset)($C) 165.6 166.7 165.3
81.4 – –
3.2.Spraydriedsamples(SD)
Twosamplesweregeneratedbyspraydryingtwodifferent
b
mannitolbatches.Thefirstonecorrespondtoapureb
mannitol obtainedbyantisolventcrystallizationandthesecondbatchtothe commercialpowderPearlitol160C. Theabbreviationsb-SD
and P160C-SDareusedinthismanuscripttodifferentiatethem. 3.2.1.PowdercrystallinityandpurityXPRDpatternsrevealedthatthetwoSDsamplesarecomposed ofthestable
b
form(Fig.4).Thesmallpeakpresentat17.3$canbeattributedtothepresence,inreallysmallquantities,of
a
mannitol. Neverthelessnotothercharacteristicpeaksfromthea
formwere found in thepatterns. Moreover,DSC and Ramanspectroscopy analysis do not corroborate thepresence ofa
mannitol in the samples.Thedifferencesbetweenthepatternspeaksintensitycan beattributedtothemorphologydifferences.SomeprotocolstospraydryD-mannitolaqueoussolutionscan
befoundintheliterature,differentiatinginpolymorphs genera-tion.Forexample,Leeetal.(2011)obtainedonlythestable
b
form afterSDwhileMaasetal.(2011)showedthatoutlettemperatures at/below90$Cgeneratedmainlyb
mannitolandsmallamountsofa
mannitol (around 5%). Outlet temperatures up to 140$Cincreased
a
mannitolconcentrationwithinthesamples(between 5 and 15%). Nevertheless, in our experiences, with an outlet temperatureof87$C,DSCandRamanspectroscopyanalysisdonotdetectedthepresenceofthe
a
formintheSDsamples.3.2.2.Particlessizeandhabitbyscanningelectronmicroscopy(SEM) ImageanalysisobtainedwithPVS830cannotbeusedforthe analysisofspraydriedsamples.Infactthemostpowerfulcamera lens “zoom 5” does not measure particles smaller than 3
mm,
whichseemstobeaquiteimportantpopulationaccordingtoSEM analysis.In addition,thedispersionprotocolusedwiththeSPD 1300 does not seem to actually disperse the particles. Finally, crystalhabitsandsizeanalysishasbeenexploitedbySEM.Froma microscopic point of view(10–20
mm),
the powders obtained after spray drying are homogeneous in size and morphology. SEM micrographs show quite spherical particles withdiametersbetween300nmand10mm
anda“quite”smooth surface (Figs. 5 and 6). These values are smaller than those obtained by Maas et al. (2011) which despite the observed differencesinparticleformation,theparticlessizedistributions obtainedatdifferentoutlettemperatures(60,90and120$C)areveryclosed, witha meandiameter of 13
mm.
The authorsalso highlightedthatthespraydryingofmannitolatdifferentoutlet temperaturesmodifiesparticlesurfacetopography.Intheircase dryingat60$Cleadstotheformationofacicularcrystalwithasmoothparticlesurface.Incontrast,athighertemperatures,the dropletsevaporatemuchfasteranddonotrecrystallize immedi-atelybecauseofthelownucleationrateatthistemperature.Evenif there is expectedthat water evaporate prior crystallization, no difference in particle size were obtained. Thus, the authors assumedthatthenucleationandcrystalgrowthalsooccursina
Fig.3.SEMmicrographsof(a)commercialpowderPearlitol160C,(b)purebmannitoland(c)pureamannitol.
10 15 20 25 30 35 40 0 1 2 3 4 5 6 7x 104 2θ Intensity β (1) Pearlitol SD (2) β−SD (3) 17.3° (2) (1) (3)
ratherearlydryingstageduetothehighdynamicinthespraydryer and the presence of crystalline seeds that may triggerearliest crystallization. In fact, since thecrystals grow larger in higher temperatures and because of the fast solvent evaporation, the formingshellbecomesquicklyveryrigidandtheopeningsofthe
hollow particles can be seen. In our case, with an outlet temperatureof87$C,someoftheparticlesexhibitaroughsurface
andsomeothershowedtobehollowwithawellvisibleorificein their shell (Figs. 5 and 6). SEM analyses were limited due to samplesdegradationforamagnificationhigherthan6400%.
Fig.5. SEMmicrographsofpurebmannitolsampleafterspraydrying.
Finally,nodifferencesinsizeandparticlesshapewerefound between
b-SD
(Fig.5)andP.160-SD(Fig.6)samples.3.2.3.Surfaceenergyanalysis
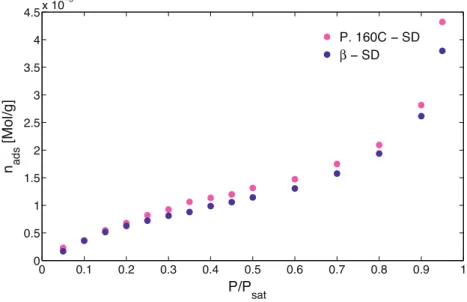
Afterspraydrying
b-SD
andP160C-SDpresentedanincrement oftheirspecificsurfaceareaandsoanincreaseofthevaporprobes adsorbedamount(Table3)allowingDVSanalysis.Thus,bothSD samples, exhibit a similar adsorption behavior with a type II isotherm(Fig.7).Surface energy values obtainedby DVS were calculatedtakingintoaccountcontactangle valuesobtainedby capillary rise (Cares et al., 2014). The error associated to the measureis1mJm!2.The
g
dsvaluesobtainedbyDVSofb-SD
andP160C-SDsamplesaresimilartothoseobtainedbyIGCforthe
b
pureform(Table3). IGC-IDanalysesshowthatb-SD
andP160C-SDsampleshave similaradsorptionbehavior(Table3).Moreover,g
ds valuesofSD
Table3
Specific surfaceareas anddispersivesurfaceenergiesofdifferentD-mannitol samples.The“X”representstheanalysisthatcannotbedone(techniquelimits) while“–”representstheonesthathavenotbeingdone.
D-mannitol as(m2g!1) gd s(mJm!2) ASAP-Ar IGC-C9 DVS IGC us=0.04% us=0.1% us=1% us=8% P160C 0.4 0.4 X 70.7 69.1 58.0 44.9 a 8.4 8.5 51 74.9 75.0 72.1 45.5 b 2.8 0.4 X 40.9 40.9 39.4 38.2 P160C-SD 3.4 4.0 40 62.6 62.3 55.4 X b-SD 2.9 4.4 41 57.2 57.3 51.7 X a-CM – 11.9 – 85.6 81.7 70.7 X b-CM 3.6 3.3 47 61.9 62.0 52.4 X 0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 0 0.5 1 1.5 2 2.5 3 3.5 4 4.5 x 10−5
P/P
satn
ads[Mol/g]
P. 160C − SD
β
− SD
Fig.7. Adsorptionisothermsofb-SDandP160C-SDobtainedbyDVSusingn-nonaneasvapourprobe.
0 0.001 0.002 0.003 0.004 0.005 0.006 0.007 0.008 0.009 0.01 30 35 40 45 50 55 60 65 70 75
n/n
mγ
s d[mJ/m
2]
β β − SD P. 160C P.160C − SDsamplesarebiggerthanthoseofthe
b
pureformbutsmallerthan those obtainedforthe commercialpowderP160C (Fig.7).This tendencyofthesamplestohavesimilaradsorptionbehaviorafter spraydryingcanbeexplainedbytheevolutionoftheparticlesto thesamesizeandcrystalshabit.Itseemsimportanttohighlight thattheg
dsofP160C-SDisapproximately5mJm!2biggerthantheoneof
b-SD
forallsurfacecoveragestudied(0.0004<n/nm<0.01). Thisdifference,superiortotheexperimentalerror(<1.3mJm!2), can be attributed to the sorbitol presence in the commercial powder P160C, which one is conserved after spray drying the sample.Itappearsthatsmallquantitiesofsorbitolinthepowders (lessthan1%)increasethestrengthofdispersiveinteractionsofthe soliddispersion.Afterspraydryingthesampleswecanfoundaregionwherethe retentiontime doesnotdependontheinjectionvolume(Fig.8, dottedlinen/nm<0.02).
3.3.Cryogenicmilledsamples(CM)
The
a
andb
D-mannitolpolymorphsgeneratedbysuccessiveprecipitationprotocolswerecryogenicmilledinordertochangeits sizeandhabitstructurewithoutchangingtheirsolid-stateform. Theabbreviations
b-CM
anda-CM
areusedinthismanuscriptto identifythecryomilledpowders.3.3.1.Powdercrystallinityandpurity
XRPD patterns were used to confirm polymorphism and to ensure the physical form of the samples.
a
mannitol has characteristicreflectionpeaksat2upositionsof13.7$,17.3$ and19.9,whilefor
b
mannitolarelocatedat14.7$,18.8$and23.6$.AsshowninFig.8,both
a
andb
puresampleskeeptheirpolymorphic formaftercryomilling.ParticlehabitandsizechangesafterCMcan bedepictedasadecreaseofthecharacteristicpeaksintensity. 3.3.2.Particlessizeandhabitbyscanningelectronmicroscopy(SEM)SEMmicrographsoftheCMsamplesareshowninFigs.10and 11. The
a-CM
samples areformedby agglomeratesof approxi-mately50mm
composedbysmallneedle-likeparticles,andalsoby agglomeratesof biggerparticles alwaysin needle-like shapeas illustratedinFig.9.Thecloserzoomsshowthatfora-CM
samples thedirectionofthefracturefollowsparticlesgeometry.Infact,it seemsthatcrystalsfracturefollowsaplaneperpendiculartothe longitudinalaxisoftheneedle.The SEM analysis of the
b-CM
samples showed particles agglomerations. The powder seems to be composed by quite irregularparticleshabitsbetweensomemicrometersand80mm.
A largequantityofsmallparticlesseemstobestickaroundthebigger ones. For this polymorph the fracture behavior aftercryogenic millingismorecomplexandthesolidseemstohaveamorebrittle behavior(Fig.11).The results obtained by image analysis using the PVS830 showedforthe
b-CM
aD[y,0.1]of8mm,
D[y,0.5]of16mm
andD [y,0.1]of30mm
whileforthea-CM
samplesaD[y,0.1]of9mm,
D [y,0.5]of24mm
andD[y,0.1]of40mm
wereobtained.NorealFig.10.SEMmicrographsofamannitolcryomilledsamples.
10 15 20 25 30 35 40 0 1 2 3 4 5 6 7 8 9 10x 104 2θ Intensity α (1) α − CM (2) β (3) β − CM (4) (3) (1) (4) (2)
differences were detected between the samples using this technique. The image analysis by SEM and by PVS830 differs, mostofallduetoparticlesagglomeration.
3.3.3.Surfaceenergyanalysis
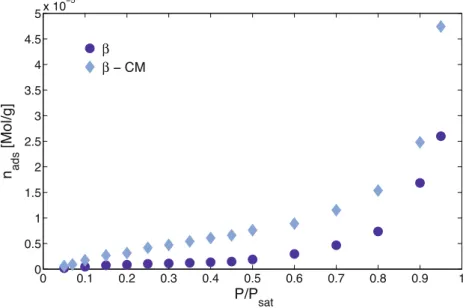
After cryogenic milling both samples,
b-CM
anda-CM,
increasedtheirspecificsurfaceareas.Thisincrementallowedto analyzebyDVStheb-CM
samplesthankstoanincrementofthe mass up-takeat reallylowpartial pressures,revealinga typeII adsorptionbehavior(Fig.12).Thispointisquiteimportant,indeed ifsolid/probe moleculesinteractionshowed atype IIIisotherm, probe–probemoleculesinteractionswillbestrongerthatprobe–solidinteractionsandsotheretentiontimemeasurebyIGCwill notberepresentativeoftheadsorptionphenomenon.
Duetothelow amount ofexperimentaldata(adsorbedamountas functionofP/Psatrangedbetween0and"0.2),between3and4,and thelowcorrelationcoefficientsobtainedwiththeDVSdata,the specificsurfaceareacalculationshavebeencarriedoutusingIGC. AsshowninTable3,anincreaseinthedispersivesurfaceenergy of
b
anda
cryogenicmilledsamples,atlowsurfacecoverage,has beenobserved.Nevertheless,forthea-CM
samplethisdifference, withthea
puresample,tendstodecreaserapidlywithincreasing theinjectedamount.Indeed,forsurfacecoveragesuperiorsthan 0.1%bothsolids,a
anda-CM,
havesimilaradsorptionbehavior.Fig.11.SEMmicrographsofbmannitolcryomilledsamples.
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 0 0.5 1 1.5 2 2.5 3 3.5 4 4.5 5 x 10−5
P/P
satn
ads[Mol/g]
β
β
− CM
The
b-CM
sample hasa similarbut more energeticadsorption behaviorthan theb
pure form.The regionwhereg
ds doesnotdependoninjectionvolumecorrespondtoaradion/nmlowerthan 0.002.Nevertheless,The
b-CM
sampleismoreheterogeneous,at low surface coverage, than theb
pure sample. This more heterogenic adsorption behavior can be attributed to the anisotropicmorphologyoftheb-CM
particles(Fig.13).4. Discussion
4.1.Henry’sdomaindeterminationbyIGC
Thefirstpointtohighlightisthataftertreatment,spraydrying and cryogenic milling, the Henry’s domain of each sample is reducedtoreallylow surfacecoverage’s(us<1%).Thus, surface energysolidanalysesdonotallowedtorepresentthewholesolids surfacebutallowedustodifferentiatebatchesbehavior.
Itshouldbenoticethatthelinearityoftheisotherm,indicated by its correlation coefficient, plays a key role over the
determination of themaximum
u
sthat define Henry’sdomain, andshouldbewelldefinedinthistypeofanalysis.Forexample,for theb-SD
samples,ifweacceptthelinearityoftheisothermfor R2>0.99,aR2=0.998correspondstoaP=40Paandsotoau
s=10% (Fig.14a)whileaR2=0.999correspondstoaP=140Paandsotoa
u
s=40%(Fig.14b).Nevertheless,wecanseethatevenifR2ishigher taking account higher pressures, the data fit used does not representquitewellthefirstpoints.Thus,inthiscase,pressures smallerthan40PaareusedtodescribeHenry’sdomain.As
g
ds isdeterminatebytheinjectionofaseriesofalkanesn-octane,n-nonaneandn-decane,Henry’sdomainwillbe determi-nate by the probe having the narrowest Henry’s domain. As expected,thelongerthechainsofalkanes,thesmallerthelinear region of theadsorptionisotherm.Sointheseexperiences, the limitsurfacecoveragetostudysurfaceenergybyIGCintheinfinite dilutionregionwillbedeterminedbytheanalysisofn-decane.
Ahighcontroloftheinjectionsizeasanimportantnumberof experimental data points it is fundamental to determine with precisionHenry’sdomain.
0 0.001 0.002 0.003 0.004 0.005 0.006 0.007 0.008 0.009 0.01 40 50 60 70 80 90
n/n
mγ
s d[mJ.m
−2]
α
α
− CM
β
β
− CM
Fig.13.Surfaceheterogeneityanalysisofaandbmannitolsamples,beforeandaftercryogenicmilling(CM).
4.2.Dispersivesurfaceenergy
Itisquitedifficulttolinkcrystalhabit,sizeandagglomeration statetosurfaceenergy.Generally,surfaceenergydifferencesafter physical treatmentsuch as milling,micronization, CM evenSD amountothers,aregenerallyattributedtochangesinparticlessize (surfacearea),morphology(habits), polymorphism(or amorph-ization)andtotheexposureofmoreenergeticallyactiveplanes. Duringourexperienceswetookcaretonotchangethesolidstate ofthesamples,thusnosolid/solidtransformationswereobtained. Moreover ithasbeendeterminedthatfor
b
mannitol,themost energeticallyactiveplane,intermsofdispersivesurfaceenergy, hasag
dsof44.1#0.6mJm!2(Hoetal.,2010).AllthisindicatesthatsurfaceenergyincrementsafterCMandSDcannotbeattributedto the exposure of new and more active phases or to solid-state transformationsaftertreatment.Thus,crystalsizeand morpholo-gychangesseemtoberesponsibleofsurfaceenergyincrementsin
b
mannitolsamples.Nevertheless,wecannotseparate particles size effectof crystalshabiteffectover thesolid surface energy because CM and SD influence at the same time these two parameters.AsshowninTable3,aftercryogenicmillingandspraydrying treatment,
a
andb
mannitolsamplespresentedanaugmentation ofitsdispersivesurfaceenergy,atreallylowsurfacecoverage.For theb
form, three batches of different size and shape were generated. Thefirstoneobtainedbyantisolventprecipitationis composed by well defined sticks with smooth surfaces D[y, 0.5]=26.7mm
andD[y,0.9]=81.7mm
(Fig.3b).Theb-SD
sampleis composedbywelldefinedspheresbetween1and4mm
(Fig.5a) while the batchb-CM
is composed byagglomerationsof quite irregular particles, of few micrometers to 90mm,
with small particlesstuckonthesurfaceofthelargerones(Fig.10).IGCanalysis revealedthatthesethreebatcheshavedifferent surface behavior. After CM and SD the samples are more energetically active due to a change in thecrystal habits. The
b-recrystallized
form appears as a quite homogeneous solid ("40mJm!2)whiletheb-CM
batchandtheb-SD
batchshoweda moreheterogeneoussurface,energeticallyspeaking.Itshouldbe notedthatdespiteitssphericalshapeandquitehomogeneoussize distribution,b-SD
samples showed a slightly heterogeneous surface(57.3–51.7mJm!2).Thisconfirmsthattheparticlessurface isnotassmoothasdepictedbySEManalysis.Surfaceenergyanalysesatreallylowsurfacecoverage,allowus todifferentiatetheCMsamplesoftheSDsamples:for
b-CM
ag
dsof61.9mJm!2whilefor
b-SD
ag
ds of57.2mJm!2at
u
s=0.04%.Theb-CM
particles are quite damaged and seem to have surface defects (Fig. 11). These surface irregularities can be seen as privilegedsiteswhere theprobemoleculescan beadsorbedby undergoing interactions from more than one surface. Surface energyanalysesat reallylowsurfacecoverageemphasizethese differences. Indeed, if the injection volume is incremented,u
s>0.1%, the surface energy differences betweenb-CM
andb-SD
samplestendtodisappearandbothsolidstendtosimilar adsorptionbehavior ("52mJm!2). Nevertheless,at this surface coveragebothsampleshaveahigherg
ds thanthedepart
b
form(52 to 40mJm!2, respectively). This difference can be still attributed to the influence of more energetically active sites related to size and crystal habit changes after SD and CM (Figs.5and11).
ItshouldbenotedthatifIGC-IDweredefinedforP/Psat"0.03,as mostauthorsdoit,wewouldworkedoutsideHenry’sdomainand wewouldalsomisleadtheeffectofparticleshabitsoversolid’s surfaceenergy.
DVSanalyses,whichprovidedameanvalueofthesolidsurface energy,showed that the
g
ds of
b-CM
samples (47mJm!2, pinkdotted line)is higher thanthe
g
ds of
b-SD
samples(40mJm!2,purpledottedline).Theseresultswithsurfaceenergyanalysesby IGC-ID showthe contributionof more active sites tothe solid averagesurfaceenergy(Fig.15).Moreover,IGC–DVSconfrontation clearlyshowsusthatifthemoreactivesitesarenotnumerous enoughornotsufficientlyhigherthanthemostenergeticactive crystalline face, its influence can be misled by the techniques whichprovidesameanvalueofthesolidsurfaceenergylikeDVS. Infact,meansurfaceenergyanalysescanmisleadtodifferentiate batchesandthusmisleadend-useproperties.
Forthe
a
form,twobatchesofdifferentsizesweregenerated but with its morphology partially conserved. Thea
mannitol generatedbysuccessiveantisolventprecipitationsiscomposedby needle-likeparticlesofD[y,0.5]=25.3mm
etD[y,0.9]=42.4mm.
Thea-CM
batch is formed by agglomerations of needle-like particles, between 20 and 50mm,
but somenon-agglomerated largerparticlescanalsobefound,alwaysneedle-like(Figs.3cand10).Surface energyanalysisshoweda moreenergeticallyactive
a-CM
sample. Nevertheless, this is only true at low surface0 0.001 0.002 0.003 0.004 0.005 0.006 0.007 0.008 0.009 0.01 35 40 45 50 55 60 65
n/n
mγ
s d[mJ/m
2]
β
β
− SD
β − CM
β
− SD
β
− CM
Fig.15.SurfaceenergyanalysisbyDVS(dottedline)andIGCofdifferentpurebmannitolbatches:bgeneratedbyantisolventprecipitation,afterspraydryingb-SDandafter cryogenicmillingb-CM.
coverage, indeed,for
u
s<0.005both samplesshoweda similar adsorptionbehavior(Fig.15).Alltheseresultscanbebetterunderstoodifthemathematical definitionoftheretentiontime istakenintoaccount.Infact,as alreadyhighlightedinpreviousstudies,tNitisthecontributionof manyinteractionssitesiandtheinteractionenergybetweenthe probeandthesitesi(Caresetal.,2014).So,ifthemoreactivesites arenotnumerousenoughitsinfluencecanbeattenuatedbythe presenceoflessactivesones,moreover,theinfluenceoflessactives siteswillbemoreimportantwhenincreasingtheinjectionsize. Thestudyofsurfaceheterogeneityathighersurfacecoveragecan giveamoregeneralideaofthispoint.Thus,for
a-CM
samples,it seemsthatthemoreactivesitescreatedbythisprocessarenot numerousenoughbecausethecryogenicmillingofa
puresamples seemstoonlygeneratesurfacedefects,whichonesseemtobeless activethanthemostenergeticallyactivefaceofthea
crystal.SurfaceenergyanalysisbyIGCatHenry’sdomainhighlighted the influence of particles habits and size over solid’s surface behavior.ThecombinedsetIGCandDVSvalidatedthisstatement. Nevertheless,itisquitedifficulttoseparatebotheffectsbecause mostprocessesaffectatthesametimesolid’shabitsandsize,even particles separation processes, such as sieving, are related to particlesmorphologies.
Fromacriticalpointofview,thestudyofsurfaceenergiesin Henry’sdomainbyIGC-ID,doesnotprovideaglobalideaofthe solidbehavior.Indeed,SDandCMsamplespresentedquitesmall linearadsorptionregion.Thus,only1%ofthesurfacecanbe“cover” tobeinHenry’sdomain(us<0.01).Sothequestionis,howfarare weofthesolid's“real”behavior;areweabletoactuallydistinguish crystallinefacetsinfluence(surfacechemistry),arethose impor-tant? It seems necessary tohave another technique of surface analysissuchasDVS,orevenmolecularmodeling.Itwillbealso interesting to study surface heterogeneity by IGC using an equipmentthatallowsahighersurfacecoverageandtouseother mathematicalmodelssuchastheenergydistributionfunction. 5. Conclusions
Powderbehaviordependsdirectlyofitsinterfacialinteractions, thusofthephysicalandchemicalcharacteristicsoftheparticlesof whichitiscomposed.Nevertheless,itisdifficulttolinkcrystalline structure,habits,particlesagglomeration state,sizedistribution and surfaceenergy. Classically, surfaceenergy incrementsafter physicalandmechanicaltreatmentsuchasgrinding,areattributed tochanges inparticlessize (surfacearea),morphology(habits), solidstatestructure(polymorphismoramorphization)and/orto theexposureofmoreenergeticallyactiveplanes.Inthisworkwe studiedtheinfluenceofparticleshabitsandsizedistributionover thesolidsurfaceenergy.Toachievethisgoal,twobatchesofpure
a
andb
mannitol,generatedbysuccessivecrystallizationprotocols, werecryogenicmilledand/orspraydried.Forthe
b
form,threebatchesofdifferentsizeandshapewere studied. The first one, obtainedby antisolvent precipitation, is composedbywelldefinedstickswithsmoothsurfaces,thesecond oneb-SD,
iscomposedbywell-definedsphereswhilethebatchb-CM
isformedbyagglomerationsofquiteirregularparticles.Itis quitedifficulttoseparatetheeffectofthesizefromtheeffectof particles habits because the two processes used, CM and SD, affectedatthesametimesolid’shabitsandsize.Moreover,even particles separation processes to study particles size, such as sieving,arerelatedtoparticlesmorphologies.IGC-IDstudieshighlightedsurfaceenergydifferencesbetween thesethree batches.Surface heterogeneity analyses, in Henry’s domain(0.0004<
u
s<0.01), showeda quite heterogeneousand more energetically activeb-CM
(61.9–52.4mJm!2) andb-SD
(57.2–51.7mJm!2)samples, and a quite homogeneous and lessactive
b
form("40mJm!2).Surfaceenergyanalysisatlowsurface coverage,u
s<0.01,allowedustodifferentiatetheSDsamplesof the CM samples. The solid size reduction, due to CM and SD, generatesanincrease ofthespecificsurfaceareaoftheb
pure powder,allowingDVSanalysisoftheb-CM
(47mJm!2)andb-SD
(40mJm!2)samples.For the
a
form, two batches of different size and a quite conservedneedle-likemorphologywerestudied.Thefirstbatch wasgeneratedbyseedingandfastcoolingwhilethesecondone,a-CM,
bythecryogenicmillingofthefirstsample.Surfaceenergy analysisatreallylowsurfacecoverage,u
s<0.005,showedamore energeticallyactivea-CM
sample.Theseresultsallowustoconcludethatparticleshabitsappear tobeamajorfactorinthesolidsurfaceadsorptionbehaviorofD
-mannitol polymorphs. Moreover, it seems that there is a “threshold”abovewhichtheinfluenceofmoreactivesites,here attributed to surface irregularities, is attenuated and different batches of a same polymorph tend towards similar adsorption behavior.Thisthresholddependsdirectlyofthevalueofthemore activesitesbutalsooftheirnumber.Ifthemoreactivesitesarenot numerous enough, or not sufficiently higher than the most energeticactiveface,itsinfluencewillbemisledbythetechniques whichprovidesameanvalueofthesolidsurfaceenergy.
IGC-IDprovideskeyinformationintermsofsurfaceenergyand appears to be a powerful tool to monitoring surface energy evolutionasfunctionofthegenerationprocessoraftermechanical orthermaltreatment.Evenifthesolidsurfaceanisotropyisweak and maybeundetectablebystandardanalytical methods,it can makethesolidmoreenergeticallyactiveandthusmorereactive. IGC-IDisabletodetectweaksurfaceenergyvariation,whichmay explain the behavior differences between batches, of a same polymorph, over time but also in atmospheres more or less moist.
Acknowledgment
InthememoryofElisabethRodier(1966–2014). References
Ahfat,N.M.,Buckton,G.,Burrows,R.,Ticehurst,M.D.,2000.Anexplorationof inter-relationshipsbetweencontactangle,inversephasegaschromatographyand triboelectricchargingdata.Eur.J.Pharm.Sci.9(3),271–276.
Brum,J.,Burnett,D.,2011.Quantificationofsurfaceamorphouscontentusing dispersivesurfaceenergy:theconceptofeffectiveamorphoussurfacearea. AAPSPharmSciTech12(3),887–892.
Cares-Pacheco,M.-G.,Vaca-Medina,G.,Calvet,R.,Espitalier,F.,Letourneau,J.-J., Rouilly,A.,Rodier,E.,2014.PhysicochemicalcharacterizationofD-mannitol polymorphs:thechallengingsurfaceenergydeterminationbyinversegas chromatographyintheinfinitedilutionregion.Int.J.Pharm.475,69–81.
Chamarthy,S.P.,Pinal,R.,2008.Thenatureofcrystaldisorderinmilled pharmaceuticalmaterials.ColloidsSurf.A:Physicochem.Eng.Aspects331(1), 68–75.
Crowley,K.J.,Zografi,G.,2002.Cryogenicgrindingofindomethacinpolymorphsand solvates:assessmentofamorphousphaseformationandamorphousphase physicalstability.J.Pharm.Sci.91(2),492–507.
Das,S.,Larson,I.,Young,P.,Stewart,P.,2009.Surfaceenergychangesandtheir relationshipwiththedispersibilityofsalmeterolxinafoatepowdersfor inhalationafterstorageathighRH.Eur.J.Pharm.Sci.38(4),347–354.
Desprez,S.,Descamps,M.,2006.Transformationsofglassyindomethacininduced byball-milling.J.Non-Cryst.Solids352,4480–4485.
Feeley,J.C.,York,P.,Sumby,B.S.,Dicks,H.,1998.Determinationofsurfaceproperties andflowcharacteristicsofsalbutamolsulphate,beforeandaftermicronisation. Int.J.Pharm.172(1),89–96.
Feeley,J.C.,York,P.,Sumby,B.S.,Dicks,H.,2002.Processingeffectsonthesurface propertiesofa-lactosemonohydrateassessedbyinversegaschromatography (IGC).J.Mater.Sci.37,217–222.
Feng,T.,Pinal,R.,Carvajal,M.T.,2008.Processinduceddisorderincrystalline materials:differentiatingdefectivecrystalsfromtheamorphousformof griseofulvin.J.Pharm.Sci.97(8),3207–3221.
Gamble,J.F.,Leane,M.,Olusanmi,D.,Tobyn,M.,Šupuk,E.,Khoo,J.,Naderi,M.,2012. Surfaceenergyanalysisasatooltoprobethesurfaceenergycharacteristicsof micronizedmaterials:acomparisonwithinversegaschromatography.Int.J. Pharm.422(1),238–244.
Hasegawa,S.,Ke,P.,Buckton,G.,2009.Determinationofthestructuralrelaxationat thesurfaceofamorphoussoliddispersionusinginversegaschromatography.J. Pharm.Sci.98(6),2133–2139.
Heng,J.Y.,Thielmann,F.,Williams,D.,2006.Theeffectsofmillingonthesurface propertiesofformIparacetamolcrystals.Pharm.Res.8,1918–1927.
Ho,R.,Hinder,S.,Watts,J.,Dilworth,S.,Williams,D.,Heng,J.,2010.Determinationof surfaceheterogeneityofD-mannitolbysessiledropcontactangleandfinite concentrationinversegaschromatography.Int.J.Pharm.387,79–86.
Ho,R.,Naderi,M.,Heng,J.,Williams,D.,Thielmann,F.,Bouza,P.,Keith,A.R.,Thiele, G.,Burnett,D.,2012.Effectofmillingonparticleshapeandsurface heterogeneityofneedle-shapedcrystals.Pharm.Res.29,2806–2816.
Imaizumi,H.,Nambu,N.,Nagai,T.,1980.Stabilityandseveralphysicalpropertiesof amorphousandcrystallineformsofindomethacin.Chem.Pharm.Bull.(Tokyo) 28(9),2565–2569.
Jones,M.D.,Young,P.,Traini,D.,2012.Theuseofinversegaschromatographyforthe studyoflactoseandpharmaceuticalmaterialsusedindrypowderinhalers.Adv. DrugDeliv.Rev.64(3),285–293.
Lee,Y.Y.,Wu,J.X.,Yang,M.,Young,P.M.,vandenBerg,F.,Rantanen,J.,2011.eof polymorphisminspray-driedmannitol.Eur.J.Pharm.Sci.44(1),41–48.
Lim,R.T.Y.,Ng,W.K.,Widjaja,E.,Tan,R.B.H.,2013.Comparisonofthephysical stabilityandphysicochemicalpropertiesofamorphousindomethacinprepared byco-millingandsupercriticalanti-solventco-precipitation.J.Supercrit.Fluids 79,186–201.
Luner,P.E.,Zhang,Y.,Abramov,Y.A.,Carvajal,M.T.,2012.Evaluationofmilling methodonthesurfaceenergeticsofmolecularcrystalsusinginversegas chromatography.Cryst.GrowthDes.12(11),5271–5282.
Maas,S.G.,Schaldach,G.,Littringer,E.M.,Mescher,A.,Griesser,U.J.,Braun,D.E., Urbanetz,N.A.,2011.Theimpactofspraydryingoutlettemperatureonthe particlemorphologyofmannitol.PowderTechnol.213(1),27–35.
Nezzal,A.,Aerts,L.,Verspaille,M.,Henderickx,G.,Rendl,A.,2009.Polymorphismof sorbitol.J.CrystalGrowth311,3863–3870.
Newell,H.,Buckton,G.,2004.Inversegaschromatography:investigatingwhether thetechniquepreferentiallyprobeshighenergysitesformixturesofcrystalline andamorphouslactose.Pharm.Res.21(8),1440–1444.
Ohta,M.,Buckton,G.,2004.Determinationofthechangesinsurfaceenergeticsof cefditorenpivoxilasaconsequenceofprocessinginduceddisorderand equilibrationtodifferentrelativehumidities.Int.J.Pharm.269(1),81–88.
Otte,A.,Carvajal,M.T.,2011.Assessmentofmilling-induceddisorderoftwo pharmaceuticalcompounds.J.Pharm.Sci.100(5),1793–1804.
Otte,A.,Zhang,Y.,Carvajal,M.T.,Pinal,R.,2012.Millinginducesdisorderin crystallinegriseofulvinandorderinitsamorphouscounterpart.CrystEngComm 14(7),2560–2570.
Pilcer,G.,Wauthoz,N.,Amighi,K.,2012.Lactosecharacteristicsandthegeneration oftheaerosol.Adv.DrugDeliv.Rev.64(3),233–256.
Planinsek,O.,Zadnik,J.,Kunaver,M.,Srcic,S.,Godec,A.,2010.Structuralevolutionof indomethacinparticlesuponmilling:time-resolvedquantificationand localizationofdisorderedstructurestudiedbyIGCandDSC.J.Pharm.Sci.99(4), 1968–1981.
Saxena,A.,Kendrick,J.,Grimsey,I.,Mackint,L.,2007.Applicationofmolecular modellingtodeterminethesurfaceenergyofmannitol.Int.J.Pharm.343,173– 180.
Shariare,M.H.,DeMatas,M.,York,P.,Shao,Q.,2011.Theimpactofmaterial attributesandprocessparametersonthemicronisationoflactose monohydrate.Int.J.Pharm.408(1),58–66.
Surana,R.,Randall,L.,Pyne,A.,Vemuri,N.M.,Suryanarayanan,R.,2003. Determinationofglasstransitiontemperatureandinsitustudyofthe plasticizingeffectofwaterbyinversegaschromatography.Pharm.Res.20(10), 1647–1654.
Tang,P.,Chan,H.,Choiou,H.,Ogawa,K.,Jones,M.D.,Adi,H.,Buckton,G.,2009. Characterisationandaerosolisationofmannitolparticlesproducedviaconfined liquidimpingingjets.Int.J.Pharm.367,51–57.
Traini,D.,Young,P.M.,Thielmann,F.,Acharya,M.,2008.Theinfluenceoflactose pseudopolymorphicformonsalbutamolsulfate–lactoseinteractionsinDPI formulations.DrugDev.Ind.Pharm.34(9),992–1001.
Ticehurst,M.D.,Rowe,R.C.,York,P.,1994.Determinationofthesurfacepropertiesof twobatchesofsalbutamolsulphatebyinversegaschromatography.Int.J. Pharm.111(3),241–249.
Thielmann,F.,Burnett,D.J.,Heng,J.Y.,2007.Determinationofthesurfaceenergy distributionsofdifferentprocessedlactose.DrugDev.Ind.Pharm.33(11), 1240–1253.
Tong,H.H.,Shekunov,B.Y.,York,P.,Chow,A.H.,2001.Characterizationoftwo polymorphsofsalmeterolxinafoatecrystallizedfromsupercriticalfluids. Pharm.Res.18(6),852–858.
Tong,H.H.,Shekunov,B.Y.,York,P.,Chow,A.H.,2006.Predictingtheaerosol performanceofdrypowderinhalationformulationsbyinterparticulate interactionanalysisusinginversegaschromatography.J.Pharm.Sci.95(1), 228–233.
Trowbridge,L.,Grimsey,I.,York,P.,1998.Influenceofmillingonthesurface propertiesofacetaminophen.Pharm.Sci.1,310.
Yamauchi,M.,Lee,E.-H.,Otte,A.,Byrn,S.R.,Carvajal,M.-T.,2011.Contrastingthe surfaceandbulkpropertiesofanhydrateanddehydratedhydratematerials. Cryst.GrowthDes.11(3),692–698.
York,P.,Ticehurst,M.D.,Osborn,J.C.,Roberts,R.J.,Rowe,R.C.,1998.Characterisation ofthesurfaceenergeticsofmilledDL-propranololhydrochlorideusinginverse gaschromatographyandmolecularmodelling.Int.J.Pharm.174(1),179–186.