O
pen
A
rchive
T
OULOUSE
A
rchive
O
uverte (
OATAO
)
OATAO is an open access repository that collects the work of Toulouse researchers and
makes it freely available over the web where possible.
This is an author-deposited version published in :
http://oatao.univ-toulouse.fr/
Eprints ID : 19415
To link to this article :
DOI: 10.1016/j.electacta.2017.01.160
URL :
http://dx.doi.org/10.1016/j.electacta.2017.01.160
To cite this version :
Lan, Yandi and Coetsier, Clémence and Causserand,
Christel and Serrano, Karine On the role of salts for the treatment of
wastewaters containing pharmaceuticals by electrochemical oxidation using
a boron doped diamond anode. (2017) Electrochimica Acta, vol. 231. pp.
309-318. ISSN 0013-4686
Any correspondence concerning this service should be sent to the repository
administrator:
[email protected]
On
the
role
of
salts
for
the
treatment
of
wastewaters
containing
pharmaceuticals
by
electrochemical
oxidation
using
a
boron
doped
diamond
anode
Yandi
Lan,
Clémence
Coetsier,
Christel
Causserand,
Karine
Groenen
Serrano*
LaboratoiredeGénieChimique,CNRS,INPT,UPSUniversitédeToulouse,118routedeNarbonne,F-31062Toulouse,FranceARTICLE INFO ABSTRACT
Refractorypharmaceuticalsremaininbiologicallytreatedwastewaterandarecontinuouslydischarged intoaquaticsystemsduetotheirlimitedbiodegradability.Electrochemicaloxidationispromisingforthe treatmentofsuchrefractorycompounds,inparticularusingaborondopeddiamond(BDD)anode.This studyinvestigatestheroleofsalts,suchassulfatesandchloridesintheelectrochemicaltreatmentof wastewater.Thepresenceofsulfatesacceleratedtheremovalofciprofloxacinandsulfamethoxazole,but hadnoeffectontheoxidationofsalbutamol.Thiscomparisonhighlightstheselectivityofthereaction betweenorganicsandsulfateradicals.Theadditionofchloridesintothesolutionledtoa remarkably-fasterdegradationofciprofloxacin.However,incompletemineralizationwasobservedathighcurrent densitiesduetothesignificantformationofhalogenatedorganiccompounds(AOX).Theformationof refractoryandtoxiccompoundssuchasClO4!andAOXcanbelimitedunderthecontrolof(i)applied
currentintensityand(ii)durationofelectrolysis.Electrochemicaloxidationofconcentrated biologically-treatedhospitalwastewaterinvestigatedtheexcellentremovalofbiorefractorypharmaceuticalsand confirmedtheaccelerationeffectofsaltsonpharmaceuticaldegradation.
1.Introduction
Recently,numerousstudieshavedemonstratedtheoccurrence of micropollutants, especially biorefractory pharmaceuticals, in theaquaticenvironment[1–4].Theyposesignificantthreatstothe sustainabilityofbothecosystemsandthesafetyofdrinkingwater
[5,6].Thesemoleculesseemstobeneitherfullyeliminatedinthe conventionalwastewater treatment plantwithactivated sludge treatment[7–9]norusingamembranebioreactor(MBR)process
[10].Consequently,analternativetechniqueisrequiredfortheir efficient elimination at source before disposal and dilution in seweragesystem.
Itis wellknownthatelectrochemicaloxidationusinga BDD anode represents a promising technique for the elimination of persistentorganics[11].Indeed,thestrongoxidationabilityofthe BDD anode is due tothe electrogeneration of hydroxyl radical ("OH)fromthewaterdischarge(Eq.1).
H2O!"OH+H++e– (1)
It is commonly assumed that electrogenerated hydroxyl radicals,themostpowerfuloxidantsinwater(standardpotential E$=2.74V/SHE[12]),areveryactiveinthedegradationoforganic molecules via the transferof oxygen atoms. Numerous studies werecarriedouttoinvestigatetheelectrochemicalbehaviorofthe BDDanodefortheremovalofpharmaceuticalproducts,suchas17
b
-estradiol[13],estrone[14],paracetamol[15],sulfamethoxazole[16,17],atenolol[18]andtrimethoprim[17]insyntheticsolutions. In all cases, total mineralization has been achieved. Moreover, coupling processes using a step to preconcentrate the micro-pollutants,followedbytheelectro-oxidationwiththeBDDanode wereperformedfor thetreatment ofrealwastewater.Urtiaga's groupcarriedoutapilotsystemthatintegratedultrafiltration(UF), reverse osmosis (RO) and electrochemical oxidation (EO) of effluents fromwastewater treatment plants [19,20].A groupof 12 pharmaceutical products was selected to monitor their degradation by the coupling processes. The treatment of the solutionby RO and EO couplingsignificantly reduced thetotal micropollutants. A previousstudy in our group performed the electrochemicaloxidationcouplingwithnanofiltration(NF)forthe treatmentof biologically-treatedhospitalwastewater(by mem-branebioreactortreatment)[21].Theresultsdemonstratedthat rapidmineralizationoccurred:theremovaloftotalorganiccarbon
* Correspondingauthor.
E-mailaddress:[email protected](K.GroenenSerrano).
(TOC)andchemicaloxygendemand(COD)reached97%and100%, respectively.Moreover,itwasnoticedthattheCODofNFretentate decayed at a faster rate than the theoretical values and the experimentalvaluesobtainedinasyntheticsolutionattheendof electrolysis.Itcanbeassumedthatadistinctoxidation phenome-nonproceedsinadditiontoEO.WithaBDDanode,itispossibleto generatestrongoxidantsfromsaltssuch assulfates,carbonates, and chlorides which are present in the solution [22]. These oxidants can act as indirectoxidants for the mineralization of micropollutantsneartheanodeand/orinthebulkofthesolution. Morespecifically,sulfateionswhichareoftenpresentinboth liquideffluentsandinnaturalwatercanbeconsideredtobean activeelectrolyte.Strongoxidantssuchassulfateradicals(SO4"!) andpersulfate(S2O82!)canbeelectrogeneratedusingaBDDanode [23]. The mechanism of the formation of sulfate radicals and persulfateis wellknown;only HSO4!and undissociatedH2SO4 reactwith"OHradicalstoformsulfateradicals[24].
HSO!
4þO H!SO
"!
4 þH2O k2¼6:9'105M!1s!1 ð2Þ
H2SO4þ"OH!SO"!4 þH2OþHþ k3¼1:4'107M!1s!1 ð3Þ TheradicalSO4"-mayproduceS2O82!byrecombination[24]. SO"!
4 þSO
"!
4 !S2O2!8 k4¼7:6'108M!1s!1 ð4Þ Recentstudieshavereportedtheinfluenceofthepresenceof sulfateontheremovalofpharmaceuticals.Murugananthanetal. haveobservedthattheeffectivemineralizationofketoprofencan only be achievedwhen the supportingelectrolytewas Na2SO4, comparingtoNaNO3,NaClandexplainedthisphenomenontothe generationofS2O82!andSO4"!fromSO42!withaBDDanode[25]. However, the actual role of sulfate and the relative electro-generatedoxidantsindegradationofpharmaceuticalproductsis not sufficient clear. Therefore, further studies are needed to investigate the role of these oxidants in the degradation of pharmaceuticals.
Thepresenceofchlorideionsallowsincreasingtheefficiencyof the degradation of organics, while at the same time, toxic perchlorateandhalogenated organiccompoundscanbeformed during the treatment using BDD anode [26–30]. Since these compoundsposeaserioushazardfordrinkingwaterandaquatic ecosystems [31,32] and are resistant to further oxidation, it is extremely important to minimize the formation of these compoundsduringwatertreatment.
PreviousworkshaveinvestigatedthatClO4!isgeneratedviaa multistepoxidationpathwayfromchloride,asshowninreaction
(5)[32].
Cl!!OCl!!ClO!
2 !ClO!3!ClO!4 ð5Þ
UsingaBDDanode,besidesdirectelectrontransferreactions, theelectro-oxidationpathwayofCl!alsoincludesthechemical oxidationreactionswithhydroxylradicals[33–39].Thepossible pathwayisexhibitedinreactionscheme(6)listedinTable1.
Cl- HOCl / OCl- ClO
3- ClO4 -a b c 6e -d 2e -e OH ClO2- f 2e -g OH h OH, e -(6)
Moreover,reactionsofadditionandsubstitutionbetweenthe organicsandtheactivechlorinespecies(e.g.Cl2,OCl-,HOCl)or chlorine radicals (Cl", Cl
2"!) are mainly responsible for the formationofundesiredhalogenatedorganiccompounds[32].
Fewstudiesdiscussaboutpossibilitytocontroltheproduction of perchlorate and halogenated organic compounds using BDD anode during the electroxidation process. Bergmann et al. has indicatedthatalowercurrentdensityandahigherflowratemay reduceperchlorateproduction,buttheseactionsalsoleadedtoa decreaseinthetreatmentefficiency[26].DonaghueandChaplin haveinvestigated that thepresence oforganics in thesolution inhibitstheformationofClO4![43].Thiswasmainlytobedueto thefactthatorganicsreactwithhigher"OHreactionratesthanthe reactionbetweenClO3"and"OHtoformClO4!(reactionh,Table1). Costaetal.havefoundthattheformationofhalogenatedorganic compoundsisfavoredin anacidicpH[27].Schmalzetal.have demonstrated that theformation of halogenated organic com-poundscorrelatedwiththeelectricalchargeanddidn’tdependon thecurrentdensity undertheirexperimental conditionsduring electrochemical disinfection of biologically-treated wastewater
[28].
Consideringthepresenceofvarioussaltsinrealwastewaterand the uncommon electrochemical properties of the BDD anode allowingthepossibleformationof perchlorateand halogenated organic compounds, a specific study is needed to suggest the appropriate operating conditions to limit the formation of undesiredandtoxicspecies.
Theobjectofthepresentstudyistoinvestigatethepositive andnegativeimpactsofthepresenceofsalts(chloride,sulfate, carbonate anions and calcium, magnesium cations) in the electrochemical treatment of wastewaters containing
Table1
Reactionsofchlorinespeciesduringelectrolysis.
Reaction Ref. a Cl! !BDD(Cl")+e! Cl"+Cl" !Cl2 Cl2+H2O!HOCl+Cl!+H 2O E0=1.36V [40,41] b Cl!+nullOH$ClOH" ! k=4.3'109M!1s!1 [42] c 6HOCl!þ3H 2O!2ClO!3þ4Cl!þ12Hþþ3=2O2þ6e! 6OCl!1þ3H 2O!2ClO!3þ4Cl!þ6Hþþ3=2O2þ6e! E0=0.46V [33] d OCl! þ2OH!!ClO! 2þH2Oþ2e! (alkalinesolution) [35] e "OH+OCl! !"ClO+OH! "OHþ"OCl! !ClO! 2þHþ k=9'109M!1s!1 k>1'109M!1s!1 [34] f ClO!
2þ2OH!!ClO!3þH2Oþ2e! (alkalinesolution) [35]
g "OHþClO! 2!"C lO2þOH! "OHþ"ClO 2!ClO!3þHþ k=6'109M!1s!1 k=4' 109M!1s!1 [34] h "ClO 3+"OH!HClO4 [39]
pharmaceuticals.Thisistheaimofproposingadequateoperating conditionstolimittheriskofformationoftoxicby-products.For that,afirstsectionperformedinsyntheticsolutionsisdevotedto thestudyoftheinteractionbetweensalts,suchaschloridesand sulfateswithhydroxyl radicalsgeneratedat the anodesurface underanodicpolarization.Theconsequencesontheremovalon three targeted pharmaceuticals: ciprofloxacin, salbutamol and sulfamethoxazolearestudied.Particularattentionhasbeenpaid on the conversion and variation in concentrations of chlorine species, as well as the formation of halogenated organic compounds. Based on these studies in synthetic solutions, electrochemicaloxidation treatmentof nanofiltrationretentate of biologically-treated hospital wastewater was performed. In viewtotheapplicationof theprocess in realwastewater, it is studiedtheeffectofthecompositionoftherealmatrixon(i)the performanceof themineralizationof pharmaceuticalproducts, (ii) the formation of by-products and (iii) the scaling on the cathode.
2.Materialandmethods 2.1.Chemicalsandsolutions 2.1.1.Chemicals
The ciprofloxacin (CIP) (+ 98% purity) was purchased from FlukaCompany.USA).Thesalbutamol(SALBU)(Salbutamol
,
sulfate +99%purity)andsulfamethoxazole(SMX)(+98%purity)were obtainedfrom Alfa Aesar and Fluka, respectively. All synthetic solutionswerepreparedwithultrapurewater(r
=18.2MV
cm). The structures of these molecules are reported in Table 2. Potassiumsulfate(+99%purity)andpotassiumchloride(+99% purity)wereanalyticalgradeandsuppliedbyFisherScience.Other chemicals,organicsorsolventswereHPLCoranalyticalgrade. 2.1.2.WastewatersourceThehospitalwastewaterwastreatedbyamembranebioreactor (MBR)installedatPurpanhospitallocatedinToulouse,France[44]. Itwasdirectlyfedfromthehospital'ssanitarycollectionsystem. MBReffluentcontainssalts,organics(totalorganiccarbonaround 20mg L-1) and around 50 pharmaceuticals from 10 different therapeutic classes [44]. Before electrochemical treatment, the MBReffluentwasfirstlyconcentratedbyaNFprocess[21].
Thebatch filtration was conductedin a cross-flowfiltration unit.Thepolyamidemembrane:NE70wasinstalledintoastainless cross-flow cell (Sepa CF II, Osmonics) in which the effective membraneareawas1.4' 10-2m2.TheNFretentatewascollected foravolumereductionfactorof5(VRF=5,correlatingtothe80% recovery)whichiswithintherangethatwouldbeappliedina full-scalesystem(30-90%).TheNFretentatewasthemixturefrom
several filtrations.The characteristicsof thewastewater matrix (NFretentateofMBReffluents)arepresentedinTable3. 2.2.Analyticaltechniques
Concentrationsofsaltsin syntheticsolutionandwastewater weremeasuredbyionicchromatographywithanICS3000system (Dionex,France).Theinjectionvolumewas25
m
Landthecolumn temperature wassetat30$C.Theconcentrations ofanionsand cations were analyzed with two columns (Thermo Scientific, Dionex):IonPacTMAS11(mobilephase:95%of5mMNaOHand5% of100mMNaOH),IonPacTMCS12(mobilephase:CH4O3S20mM), respectively.Analyticalerrorsforanionsandcationsrangedfrom 1.5%(Cl!)to6%(SO
42!).Inthismobilephase,HClOwasreportedas ClO!.
Pharmaceuticalconcentrationsweremeasuredbyhigh perfor-mance liquid chromatography connected with an ultraviolet-visible spectrometry detector (HPLC-UV). The analyses were conductedonAgilent1200SeriesHPLCsystems(Agilent Technol-ogies,USA).AnAgilentZORBAXEclipsePlusC18column(3.5
m
m, 3mm'100mm) from Agilent Technologies was used. The detectionUV wavelength was setto278nm.The mobilephase ofHPLCconsistedofagradientofultrapurewater(with0.1%formic acid) and acetonitrile (with 0.1% formic acid) and the column temperaturewassetto30$C.Theflowratewas0.4mLmin-1and the volume of injection was 10m
L. The detection limit of ciprofloxacin and sulfamethoxazole is 10m
gL-1 and 100m
gL-1 forSalbutamol.Theanalyticalerrorsrangefrom0.2%to1%.Adsorbableorganichalogens(AOXs)wereanalyzedin accor-dancewiththeISO9562Method[45].ISO9562usuallyspecifies themethodologyforthedirectdeterminationofaconcentrationof 10
m
g L-1 in water for organochlorine, bromine and iodine (expressedaschloride)adsorbableonactivatedcarbon.TOCandinorganiccarbon(HCO3!inexperimentalconditions) were measured with a TOC-VCSN instrument (Shimadzu). The concentrationofinorganiccarbonwasmeasuredafteracidification
Table2
Structureoftargetpharmaceuticals.
Ciprofloxacin(CIP) Salbutamol (SALBU) Sulfamethoxazole (SMX) Formula C17H18FN3O3 C13H21NO3 C10H11N3O3S Molecularweight (gmol-1) 331.3 239.3 253.3 Structure Table3
Physico-chemicalcharacteristicsandcompositionofthewastewaterafterNF. Propertiesandtargetpharmaceutical Value Ions Concentration
(mgL-1) pH 7.84 Na+ 161 Conductivity(mScm-1) 1.2 K+ 34 COD(mgL-1) 86 Mg2+ 9 TOC(mgL-1) 40 Ca2+ 70 UV254 1.13 Cl- 70 AOX(mgClL-1) 2300 NO 3- 152 SO42- 110 Targetpharmaceutical PO43- 13 Ciprofloxacin(mgL-1) 81 HCO 3- 63
anddegassing,performedautomatically.TOCwascalculatedfrom thedifferencebetweenthetotalcarbonandinorganiccarbon.COD was determined by photometry using disposable test tubes (HI93754H-25 LR from HANNA Instruments) and a HACH DR/ 2400photometer.Testtubeswereheatedat150$Cfor2hoursand lefttocooldownatroomtemperaturebeforemeasurement.The analyticerrorsforTOCandCODwereestimatedto5%.
2.3.Electrochemicalset-up
The experimental solutionwas stored in a thermoregulated glassreservoir(1)andcirculatedthroughtheelectrochemicalcell usingacentrifugalpump(2)(Fig.1).Theflowratewas360Lh-1 and the volume of the solution was 1L. Electrolyses were conductedat30$Cinaone-compartmentflowfilterpressreactor undergalvanostaticconditions(3).Electrodesweretwodiscsof 69cm2 of active surface. The BDD anode from Waterdiam (Switzerland)waselaboratedbychemicalvapordepositionona conductivesubstrateofsilicium. Thecathodewasa 1mmthick discofzirconium.ThecurrentwassuppliedbyanELCAL924power supply.ThemasstransfercoefficientcanbedeterminedbyEq.7in theoperatingrangeofflowrate(120-300Lh-1)at30$C[46]: kd'105¼0:0051
F
þ0:4367 ð7Þwhere,kdisthemasstransfercoefficient(ms-1),
F
istheflow rateinLh-1Inthepresentstudy,themasstransfercoefficient correspond-ingtotheflowrateof360Lh-1equals2.30'10-5ms-1.Beforeeach electrolysis, the workingelectrodes were anodically pretreated (40mAcm-2for30minina0.1MK
2SO4solution)tocleantheir surfacesofanypossibleadsorbedimpurities.Thenthesystemwas rinsedbyultrapurewater.Samplesweretakenatregularintervals inthetank.Theglobalvolumeofsampleswaslessthan10%ofthe totalvolume.
AccordingtothepropertiesoftheBDDanode,limitingcurrent densitycanbedefinedbyusingaglobalparameter,theCOD[47].
ilim¼4FkdCOD ð8Þ
whereilimisthelimitingcurrentdensityforthemineralization oforganics(Am-2),FistheFaradayconstant(Cmol-1)andCODisin molO2m-3.
Dependingonthevalueofthecurrentdensity(i),twodifferent kinetic regimes can be defined:(1) i<ilim: the kinetics of the reactionischargecontrolled;(2)i>ilim:thekineticsofthereaction is controlled bymass transfer.In this paper, theoxidation was undermass transfercontrol(i>ilim), thus theevolutionof COD withtimecanbeexpressedasequation(9):
CODðtÞ¼COD0exp !Akdt V ! "
ð9Þ whereV andAare thevolume ofthesolution(m3)andthe electrodesurface(m2),respectively.
Inallelectrolyses presentedinthis paper,theoxidationwas under mass transfer control and for [CIP]0=0.0695mM, the correspondingi$
lim=1.29mA. 3.Resultsanddiscussion
3.1.Electrochemicaloxidationofpharmaceuticals
Fig. 2 shows the decay of pharmaceuticals (ciprofloxacin, salbutamol or sulfamethoxazole) during the electrolysis. The completeremovalofthesepharmaceuticalsisobservedataround 250min.
The comparison of the three pharmaceuticals shows an exponential decay according to the process limited by mass transfer(i>ilim)(Eq.8).Theremovalofpharmaceuticalsfollowsa pseudofirst-orderreaction(Eq.10).Thekineticanalysisisshown intheinsetpanelofFig.2.
lnCt
C0¼!kobst ð10Þ
wheretistheelectroxidation time;C0 and Ct aretheinitial concentrationofpharmaceuticalcompoundandconcentrationat time t, respectively; kobs is the observed degradation rate of pharmaceuticals.
Thedegradationrateofthethreepharmaceuticalsarealmost thesame(0.0203min-1,0.0208and0.0168min-1,for CIP,SALBU andSMX,respectively)withaslightdifferencewithSALBU.
Itiswellknownthatelectrogeneratedhydroxylradicalsreact massivelywithorganicswithoutselectivity.Thesereactionsoccur veryclosetotheBDDanode.Themainpathwaysof"OHreactions
Fig.1.Discontinuousprocesswithasinglecompartmentelectrochemicalreactor, (1)atank,(2)apump,and(3)anelectrochemicalcell.
Fig.2.Concentration variationof~ciprofloxacin (CIP),^salbutamol (SALBU), &sulfamethoxazole(SMX)duringelectrolysisin1LofK2SO40.02molL-1.Operating
conditions:[SALBU]0=0.0671mmolL-1,i=7.25mAcm-2;[SMX]0=0.0596mmolL-1,
i=7.25mAcm-2;[CIP]
0=0.0695mmolL-1,i=1.45mAcm-2.Insetpanel:kinetic
withpharmaceuticalsare additionstoC-C, C-NandC-S double bonds and H-abstraction [33]. The kinetics of most addition reactionsareveryhigh, typicallyin theorder of109-1010M-1s-1 [48,49].Consequently,thedegradationofpharmaceuticalsunder masstransfercontrolisnotselectiveandmainlydependsonthe masstransfercoefficient.
3.2.Effectofsulfateionsonpharmaceuticalselimination
Asmentionedpreviously,onlyHSO4!andundissociatedH2SO4 reactwith"OH radicals toformSO
4"!and S2O82!(Eq.2-4). To investigate the role of sulfate species on the removal of pharmaceuticals, theresults obtainedin Fig. 2 were compared withthe onesobtained in K2SO4 0.1M(pH=3.7 adjusted with H2SO40.1Msolution).InK2SO4solutions,sulfateanions, hydro-genosulfate anions and undissociated sulfuric acid coexist, dependingonthepH.Theconcentrationofeachspecies canbe calculatedfromthedissociationconstantsofEq.11and12[50]. H2SO4+H2O!HSO4-+H3O+ ka1=2.4' 106 (11)
HSO4!+H2O!SO42-+H3O+ ka2=1.0'10-2 (12) Table4presentstheconcentrationsofHSO4!andSO42!andthe pharmaceuticals in both solutions. The main form of sulfate is SO42-, the concentration of undissociated H2SO4 is negligible. Moreover,itcanbeobservedthattheconcentrationofHSO4!ina K2SO40.02Msolutionis2000timeslowerthaninaK2SO40.1M pH=3.7.Consequently,theproductionofSO4"!andS2O82!isless significantinK2SO40.02Msolution.
Fig.3 showsthedegradationofSALBU,SMXand CIPinboth electrolytes.Itisinterestingtoobservethattheeffectofelectrolyte compositiondependsonthenatureofthepharmaceuticals.
ThesamedegradationrateofSALBUwasobserved(Fig.3(a))in bothelectrolyteswhatevertheconductivityofthesolution(4.7mS cm-1 for [K
2SO4]=0.02M and 20 mS cm-1 for [K2SO4]=0.1M). However,greaterdegradation ratesof SMX (Fig.3 (b)) and CIP (Fig. 3 (c)) wereobtainedin K2SO4 0.1M, which indicates that another phenomenon occurred in both electrolyses. Additional chemical reactions between organics and electrogenerated oxidantsfromhydrogenosulfate anions, such assulfate radicals or persulfate anions (Eq. 3 and 4) can explain these results. Thermodynamically,SO4"!(E$=2.6V)isastrongeroxidantthan S2O82!(E$=2.01V).SO4"!canreactselectivelyandrapidly with pharmaceuticalsthat areclosetotheanodesurface,withinan orderof109-1010M-1s-1[51],whilethereactionofS
2O82!with manyorganicsiskineticallyslow[52].Moreover,itwasreported thatS2O82!wasnot capableofoxidizingCIP and SMXwithout activationtoSO4"![53].Therefore,theSO4"!playsadominantrole ontheadditionaloxidationofpharmaceuticalsinthepresenceof hydrogenosulfate.
In general, SO4"! is more likely to participate in electron transfer reactions [49,51,54] than "OH which is more likely to participateinhydrogenabstractionoradditionreactions[55,56]. Theformer reactionsareselectivewhilethelateronesare
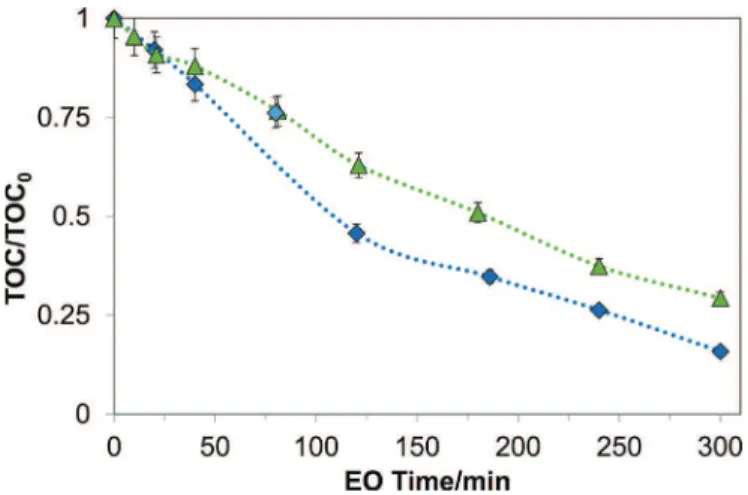
non-selective. The acceleration phenomena were observed in the degradationofSMXandCIPbutnotinSALBU,whichexclusively correlateswiththefactthatSO4"!reactsselectivelywithorganics. Overall, the electrogenerated SO4"! also accelerated the removalofTOC(Fig.4)andCOD(notshown).Resultsshowthat 84% of TOC and 100% of COD was removed in a K2SO4 0.1M solution,whileinaK2SO40.02Msolution,only71%ofTOCand86% ofCODwasremovedafter300minofelectrolysis.
3.3.Electrochemicaloxidationofpharmaceuticalsinthepresenceof chloride
ToinvestigatetheroleofCl! duringelectrolysis,besides the pharmaceuticaldegradation,itisimportanttostudythe conver-sionofchlorinespeciesandtheformationofAOXs,whichcouldbe
Table4
ConcentrationofSO42-,HSO4!andpharmaceuticalsinsyntheticsolutionsatinitial
time. Concentration(molL-1) [K 2SO4]=0.1molL-1 pH=3.7 [K2SO4]=0.02molL-1 pH=6.4(initial) [SO42!] 9.8'10-2 2'10-2 [HSO4!] 1.9'10-3 8'10-7 [SALBU] 6.77'10-5 6.71'10-5 [SMX] 6.23'10-5 6.95' 10-5 [CIP] 6.23'10-5 6.95' 10-5
Fig.3.Thenormalizedconcentrationofpharmaceuticalsduringelectrolyses:(a) SALBU,i=7.25mAcm-2;(b)SMX,i=7.25mAcm-2;(c)CIP,i=1.45mAcm-2in
K2SO40.1molL-1,pH=3.7; K2SO40.02molL-1,pHrange=6.4-4;i>i0lim.Error
apotentialenvironmentalhazard.TherangeoftheCl! concentra-tion in a synthetic solution was chosen according to the concentrationrangeintheNFretentate(Table3).
3.3.1.OxidationofCIPandCl
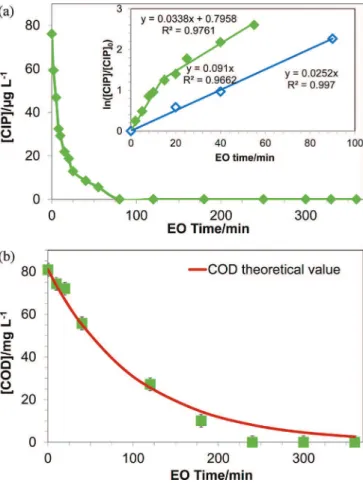
-Fig. 5 (a)showsthedegradation ofCIP in thepresence and absenceofchloridesforthespecificappliedcurrentdensitiesclose tothelimitingcurrentdensity.
TheobviousaccelerationforCIPdegradationisobservedinthe presenceofCl-:thecompleteremovalofCIPwasreachedat80min inthepresenceofCl!while240mininabsenceofCl!.Moreover, thepresenceofCl!alsoposedasignificantpositiveeffectonthe removalofTOC(Fig.5(b)).Afteranelectrolysistimeof300min, 90%ofTOCremovalwasreached,comparedtoonly70%without Cl!ions.Theaccelerationeffectofthepresenceofchlorideionsin thisstudycorrelatedwiththeelectrogeneratedactivechlorine(Cl2, HClO or ClO!) which can indirectly oxidize the organics [13,14,57,58].Moreover,duetotheelectrochemicalpropertiesof BDD, chloride radical (Cl") is the
first-step product of direct oxidation ofchloridewithBDDanode[40](Table1,reactiona). Besides active chlorine, additional inorganic oxidants such as SO4"!canbeformedfromthereactionbetweenCl"andSO42![59] (Eq.13). Thisradicalalsoplaya positiverolein theoxidation of organics.
Cl"þSO2!4 !SO"!
4 þCl! k11¼2:5'108M!1s!1 ð13Þ The effect of the presence of Cl! on mineralization of pharmaceuticals seems to be surprisingly adverse at a high applied current density. It has been found that TOC removal reached90%inthepresenceofCl!at1.45mAcm-2(i-i0
lim),while decreased dramatically to 40% at 43.5mA cm-2 (i
i0lim
>30). The efficiencyofmineralizationhighlyrelatedtotheappliedcurrent density. LowremovalofTOC maybeattributedtothepossible formationofhalogenatedorganiccompoundsduringelectrolysis. Toexplainthissurprisingeffect,afurtherstudyinthenextsection focusesontheformationofhalogenatedorganiccompoundsand theconversionofchlorinespecies.
3.3.2.ConversionofchlorideandformationofAOXs
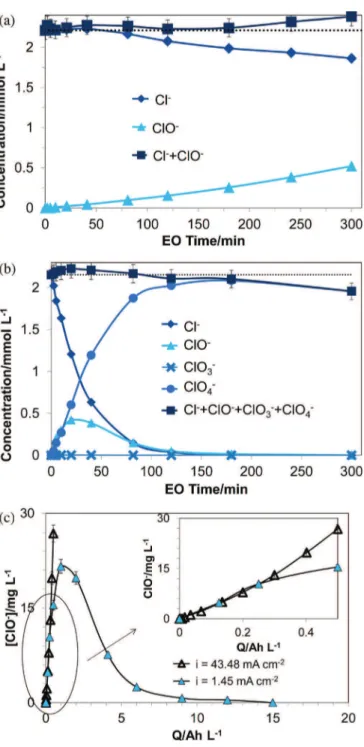
To understandtheconversionofchlorideduringelectrolysis,
Fig. 6 shows the variation of concentrations of all identified chlorinespecies(Cl-,HClO/ClO-,ClO
3-,ClO4!)withelectroxidation time at 1.45 and 43.5mAcm!2. The formation of hypochlorite species (HClO and ClO!) occurred during electrolysis at both current densities and related totheelectric charge (Fig.6 (c)).
Whenthecurrentdensitywasclosetothelimitingcurrentdensity (i=1.45mAcm-2),Cl!wasoxidizedinhypochloriteandnoother specieswasobserved(thesumoftheconcentrationofCl!andClO! equaled the initial concentration of Cl!). At higher current intensities(43.5mAcm-2), ClO! and ClO
4! wereformed atthe beginning of electrolysis. The concentration of ClO! reached a maximumvalueof0.42mMat20min,thendecreasedto0.05mM at 120min and disappeared at the end of electrolysis. The concentrationof ClO3! duringelectrolysis at 43.5mA cm-2 was lower than the quantification limit of ionic chromatography (<1ppm). After 300min of electrolysis, 90% of chloride was convertedintoperchlorate,whichwasthemajorchlorinespecies inthesolutionat43.5mAcm-2.
ItisevidencedthattheformationofClO4!ishighlyrelatedto theappliedcurrentdensity,ratherthantheelectriccharge.Since, foranelectricalchargeof0.5AhL-1,aquarterofCl!wasremoved and halfof it wasconverted intoperchlorateat a highcurrent density(43.47mAcm-2);at1.45mAcm-2,allthedisappearedCl! wereintheformofClO!.Thisphenomenoncanbeattributedto the fact that more oxygen species (mainly "OH) are electro-generated for high applied current densities. It is known that electrogenerated oxygen species play an important role in the generationofClO4!fromCl. Azizietal.haveshownthatontheBDDanode,theHClO4isformed bythehomogeneousreactionbetweenClO3"and "OH [39].The highconcentrationofelectrogeneratedoxygenspeciesallowsthe reactionsbetweentheintermediatechlorinespeciesandoxygen speciestoformClO4!.Thisexplanationisalsosupportedbythe studiesofJungetal.[36]showingthattheelectrogenerationrateof ClO4! depended on the concentration of oxygen species ("OH,
Fig.4. NormalizedvariationofTOCduringelectrolysisof1LofCIPsolution.^K2SO4
0.1molL-1,initialpH=4,[TOC]
0=10.4mgL-1,~K2SO40.02molL-1,initialpH=6.4,
[TOC]0=11.2mgL-1;i0lim=1.45mAcm-2(i>i0lim),errorbars=5%.
Fig.5. VariationofCIP(a)andTOC(b)in^K2SO40.02molL-1,&K2SO40.02molL -1+2.25mmolL-1KCl,duringelectrolysisof1LCIPsolution,[CIP]
0=22.30mgL-1
(0.068mmolL-1),[TOC]
0=12mgL-1,i=1.45mAcm-2(i0lim=1.29mAcm-2),initial
H2O2,O3).AfastergenerationrateofClO4!wasobservedforhigher concentrationsofoxygenspecies.
Moreover, the presence of organics, such as CIP which are scavengers of "OH may significantly limit the production of perchlorate.Theobviousdecreaseinchlorideconcentrationsonly occurredafterapproximately40minofelectrolysis,when90%of CIP was removed at 1.45mA cm-1. Conversely, higher current densities(43.47mAcm-1),chlorideisoxidizedfromthebeginning oftheelectrolysis.
Itisinterestingtofindthatintermediatespecies,suchasClO2! and ClO3! werenot observedbeforethe formationof ClO4!at 43.5mAcm-1. Thisis probablydue tothe electrochemicalBDD anode'spropertiesandthehighappliedcurrentdensity.Radical sites(forexampleC-O"andC")ontheBDDanodesurfaceactas adsorptionsitesforClOx"radicalsonanodesurfaces[32].Radical sitescanstabilizetheClOx"radicalsandincreasetheirlifetime, leading to favor their reaction with electrogenerated oxygen species(mainly"OH)intheareaclosetotheanode.Therelatively largeconcentrationofhydroxylradicalsforahighcurrentdensity makeitpossibletodirectlyformtheterminalproduct(ClO4!).
Furthermore,itcanbeobservedfromFig.6(b)thatthefinal concentrationofthechlorinespecies(ClO4!)waslowerthanthe initialconcentrationofCl!.TheriskofformingAOXswhichare toxictotheenvironment and human health, seemstoexist,in particularforhighcurrentintensities.Table5highlightsthatfora currentdensityof43.5mAcm-2,thequantityofAOXsattheendof electrolysisis13timeshigherthanthatforacurrentdensityof 1.45mAcm-2.
Addition and substitution reactions between organic com-poundsandactivechlorine(e.g.,Cl2,OCl,HOCl)orchlorineradicals (Cl",ClO
x")aretheprincipalreasonsfortheformationofAOXs[32]. At high current density (43.5mA cm-2) the amount of these electrogenerated chlorinespecies is veryhigh. Inparticular the chlorineradicals,whichcanbeformedontheelectrodesurface (equationb,e,g,Table1)andalsopossiblyinthebulkwiththe electrogeneratedoxygenspecies(e.g."OH,H
2O2).Forexample,in the bulk, ClO! reacts with electrogenerated H
2O2 to form a hypochloriteradicalasEq.14and15[61,62].
OHþOH!H2O2 ð14Þ
OCl!þH2O2!O ClþO HþOH!
ð15Þ Consequently, using a highcurrent density (i
i0lim
>30) in such electrochemicalprocessesposestherisksoftheformationofAOXs. Furthermore,the significantformation of AOXat 43.5mA cm-2 explainsthelowremovalofTOC(40%)attheendofelectrolysis. ThisTOCislikelytoberatherrefractorytotheelectroxidation:the valueis stablefrom120mins’(6AhL-1)to300min(15AhL-1) electrolysis.
Thus,thechoiceoftheappropriatecurrentdensity(i-i0lim)is important to limit the formation of refractory and toxic com-pounds such as ClO4! and AOXs for theapplication of electro-chemical oxidation with the BDD to treat wastewater in the presenceofchloride.
Fig.6.ConcentrationofchlorinespeciesduringelectrolysisofK2SO40.02molL-1
and2.25mmolL-1KClsolutioncontainingCIP:Cl!(^);Cl!+ClO!+ClO 4!(&);(b)
ClO!(~),ClO
3!( )ClO4!(*)(a)i=1.45mAcm-2,(b)i=43.48mAcm-2,(3)ClO!
versustheelectricalcharge,initialpH=6.Thedashedlineisthetotalconcentration ofchlorinecompoundsinthesolution.Errorbars=.1.5%.forCl-,.2%.forClO4-,.5%. forClO!.
Table5
ConcentrationofAOXatinitialtimeandafter300minofelectrolysis.Operatingconditions:Fig.6. [Cl!]
0
(mgL-1) Currentdensity(mAcm
-2) EOtime(min) Q
(AhL-1) TOC(mgL-1) AOX(mgL-1Cl)
78.0 0 0 0 12 <LD
78.5 1.45 300 0.5 1.1 370
3.4.Electrochemicaloxidationtreatmentofrealwastewater 3.4.1.Effectofmatrixontheremovaloftargetpharmaceuticalsand COD
ElectrolysiswascarriedoutwithNFretentateof biologically-treatedhospitalwastewater.Thephysico-chemicalcharacteristics andcompositionofthewastewaterarepresentedinTable3.Fig.7
shows the variation of concentrations of CIP and COD in wastewaterduringelectrolysis.
TheCIPwastotallyremovedafter80min(0.4AhL-1).Itcanbe observedthatduringthefirst15minutesCIPdegradedfastina higherrate,thereafterthedegradationratedecreased.Thechange inthedegradationratewaspossiblyinducedbytheproductionof several kindsofintermediates.ThesemaycompetewithCIP for oxidationandslowdowntheobservedCIPdegradationrateinthe complexmatrix.Nonetheless,thedegradationrateofCIPintheNF retentate matrix (0.091min-1 for the first 15min and then 0.034min-1)was higherthan theone obtainedinthesynthetic solution (performed under the same applied current density): 0.028min-1.Itcanbesuggestedthatsomeadditionalphenomena occurredduringtheelectrochemicaloxidationofwastewater.As discussedinsection3.2,theoxidantselectrogeneratedfromsalts, suchasSO4"!andhypochloritecanacceleratethedegradationof CIP.ThesimilarfasterremovalofCODwasobservedafter120min ofelectrolysis(Fig.7(b)).Thecalculationoffaradicefficiency(ICE) asfunctionofCODvaluesshowsthatICEdecreasesfrom44%to10% between10and 180minutesofelectrolysis.Atthebeginningof electrolysis,thedecayofCODwasin excellentaccordancewith theoretical values. After 120min the difference between the
experimentalandthetheoreticalvaluescanbeexplainedbythe effectofthereactionwiththeelectrogeneratedoxidantsfromsalts. 3.4.2.Conversionofions(inparticularCl!)duringelectrolysis
ThepresenceofCl!intheprocessmustbestudiedcarefully, particularlytothepossibleformationoftoxicby-products.Fig.8
presents the conversion of chlorine species during 360min electrolysisof1LNFretentate.Itappearsthattheconcentration ofCl!declinedwithtimeandthatClO!andClO
4!wereformed (concentrationofClO3!waslowerthanthequantificationlimits, thatare<1mgL-1).Thiselectrolysiswas performedwitha real wastewaterwhichcontainsmanyorganicsincludinghalogenated organics. Thus, the initial quantity of chlorinated species is unknown. During the electrolysis, the oxidation of such com-poundsmayproducechlorideionsorperchlorate.Consequently,a completemassbalanceofchlorinatedspeciescannotbeachieved. Whentheapplied current density was closetothe limiting current(i=4.34mAcm-2,i0
lim=2.25mAcm-2),thegenerationof undesiredperchlorateClO4!hasbeenobservedonlyafter180min. Atthistime,thetargetpharmaceutical(e.g.CIP)wascompletely degradedand86%ofCODwasremoved.TheClO4!appearedonly after120minutesandreached3mgL-1attheendofelectrolysis.As thoughthatthepresenceoforganicsintheNFretentateinhibited theformationofClO4!atthebeginningofelectrolysis,thenthe inhibitioneffectweakenedwiththedecreaseinorganic concen-tration. This explanation is in accordance with the study of DonaghueandChaplin[43] inwhich theorganicsactedas"OH scavengersandcompetewithClO3",whichwastheintermediateof ClO4! formation. In other words, based on Eq. 8, the limiting current density decreases with COD decay. Herein, at 180min ( i
ilimð180Þ>26),asdiscussedinsection3.3.2,theuseofhighcurrent densityfavorstheformationofClO4!.Consequently,theapplied current density, but also the electroxidation time are very significantforlimiting oftheClO4!formation.Anotherpossible strategywouldbetoadjustthevalueoftheappliedcurrentdensity withtheCODvalue,accordingtoEq.8.
Inaddition,itisworthnotingthat80%ofAOXspresentintheNF retentate(initialAOX=2300
m
gClL-1andfinalAOX=450
m
gClL-1) wereremovedafter360minofelectrolysis.Regardingtheothermineralsalts,(figureisgivenin Supple-mentaryMaterialsS1),ononehand,theconcentrationofNO3!and SO42!increasedslightlyduringtheprocessandstabilizedwhich canbeexplainedbythedegradationoforganicscontainingatoms of S and N. On the other hand, one can observe a significant decreaseintheconcentrationsofMg2+,PO
43-,Ca2+andHCO3!.This decreasecanbeattributedtothescaling,whichoccurredonthe
Fig.7.RemovalofCIP(a)andCOD(b)withelectrochemicaloxidationtimeduring 360mins’ofelectrolysisof1LNFretentate.[CIP]0=76mgL-1(0.023mmolL-1),
COD0=81mgL-1,i=4.34mAcm-2(i0lim=2.25mAcm-2),pH=8.Insetpanel:kinetic
analysisofpharmaceuticalsduringEOundermasstransfercontrolinNFretentate (fullsymbols)andinsyntheticsolution(emptysymbols).
Fig.8.Variationofconcentrationsofchlorinespeciesduring360mins’electrolysis of1LNFretentate:Cl!(^);(b)ClO!(~),ClO
3!(&)ClO4!(*),pH=7.8,[Cl!]0=2
electrolysis cell.Indeed,during electrolysis,theelectrochemical reductionofdissolvedoxygenandwaterleadstotheincreaseof thelocalpH.Thegenerationofhydroxylionsdisturbsthe calco-carbonicequilibriumofthesolution.Hydrocarbonateions(HCO3!) areconvertedintocarbonateions(Eq.16):
HCO!
3þOH!!CO2!3 þH2O ð16Þ
Carbonateionsmayreactwithcalciumionstoformcalcium carbonateonthecathodesurface.
Ca2þþCO2!3 !CaCO3# ð17Þ Inthesameway,theprecipitationofMg(OH)2happensatthe cathodes[63].
Mg2þþOH!
!MgðOHÞ2# ð18Þ
Toverifythishypothesis,theelectrochemicalcellwaswashed withpurewaterandthenrinsedwith0.7LofH2SO40.1Msolution foronehour. Theacidicsolutionwasanalyzedbeforeandafter washing.TherateofrecoveryforMg2+,PO
43-,Ca2+afteracidified rinsewasrelativelysatisfactory,withanaccuracyof20%.Evenif scalingoccurredduringelectrolysis,theincreaseincellpotential wasnotobservedduringelectrolysis.Thedepositofscalingshould beporousandshouldnothindertheelectrontransfer.However, thelong-termeffectshouldbeshorteningtheservicelifeofthe electrodes.ThesatisfactoryrateofrecoveryofMg2+,PO
43-,Ca2+ indicatesthatasimplerinsewithacidicsolutioncanbeaneffective methodtodealwiththescalingofthecell.Regularrecirculation withan acidic solution in the electrolysis system could be an efficientstrategyforprotectingtheelectrode.Anothertreatment consists of regularly applying reversal of electrode polarity, in ordertolimitelectrochemicallydissolvethedepositionofscale. 4.Conclusion
Beyond the increase in conductivity of the solution, the presenceofsaltsinthesolutiongenerallyinducesanincreasein theremovalrateofpharmaceuticals,thechemicaloxygendemand andtotalorganiccarbon.Strongoxidantscanbeelectrogenerated fromsaltsduringelectrochemicaloxidation;theseoxidantsenable to react chemically with organics. This is particularly true for chlorideandsulfate anions.Thelatterin contactwithhydroxyl radicalsformsulfateradicals thatreactselectivelywithorganic compounds:thepresenceofsulfatesacceleratedtheremovalofCIP and SMX, but had no effect on the oxidation of SALBU. A remarkableaccelerationofCIPdegradationwasalsoobservedin presenceofCl!.However,electroxidationathighcurrentdensities surprisinglyhadadverseeffectonthemineralizationof pharma-ceuticals.TOCremovalindicatedinpresenceofCl!ati
-i0limfrom 70%to90%,whiledramaticallydecreasedto40%when i
io
lim>30.The significantlyquantityofAOXsformedathighercurrentdensities canexplainthelowremovalrateofTOC.Theformationofother undesiredby-products:ClO4!wasonlyobservedathighcurrent densities(i
iolim>30).TheformationofClO4
!andAOXsisstrongly related to the applied current density rather than the electric charge. This can be explained by the fact that, under these operatingconditions, manyhydroxyl radicals areproduced and consequently,manyoxygenatomsaretransferredtothechlorine speciesandorganicstoformperchlorateandAOXs.Thistendency has been confirmed during electrolysis of real wastewaters. Perchlorate appeared when the organic matter has nearly completelydegraded.
Theseresultshighlightthatitisveryimportanttocontrolthe electrolysistimeinordertolimitperchlorateformationortoadapt
the value of the applied current density with the COD value accordingtothetheoreticalmodelofCODdecay.
Moreover, the presence of Ca2+, Mg2+ and inorganic carbon entailsscalingwhichisduetotheincreaseofthelocalpHatthe cathode.Aregularrinsingoftheelectrochemicalcellwithacidic solution oran electrochemical treatmentconsisting in reversal polarity can be proposed to limit the decrease of lifetime of electrode.Thecleaningfrequencyshouldbeadjustedaccordingto thehardnessofthesolution.
Acknowledgment
TheauthorswouldliketothanktheChinascholarshipcouncil forthefinalsupport.Theyarealsogratefultothecoordinatorofthe PANACEE project supported by the French National Research Agencyforallowingusaccesstohospitalwastewater.Theywould liketothanktheLaboratoireDépartemental31: Eau-Vétérinaire-AirinToulousefortheanalysisofAOXs.Lastly,theywouldalsolike tothankSophieChambers forproofreading andcorrecting the manuscript.
AppendixA.Supplementarydata
Supplementarymaterial relatedtothis articlecan befound, intheonlineversion,athttp://dx.doi.org/10.1016/j.electacta.2017. 01.160.
References
[1]E.Zuccato,D.Calamari,M.Natangelo,R.Fanelli,Presenceoftherapeuticdrugs intheenvironment,TheLancet.355(2000)1789–1790.
[2]D.W.Kolpin,E.T.Furlong,M.T.Meyer,E.M.Thurman,S.D.Zaugg,L.B.Barber,H. T.Buxton,Pharmaceuticals,Hormones,andOtherOrganicWastewater ContaminantsinU.S.Streams,1999!2000:ANationalReconnaissance, Environ.Sci.Technol.36(2002)1202–1211.
[3]X.Chang,M.T.Meyer,X.Liu,Q.Zhao,H.Chen,J.Chen,Z.Qiu,L.Yang,J.Cao,W. Shu,Determinationofantibioticsinsewagefromhospitals,nurseryand slaughterhouse,wastewatertreatmentplantandsourcewaterinChongqing regionofThreeGorgeReservoirinChina,Environ,Pollut.158(2010)1444– 1450.
[4]K.S.LeCorre,C.Ort,D.Kateley,B.Allen,B.I.Escher,J.Keller, Consumption-basedapproachforassessingthecontributionofhospitalstowardstheloadof pharmaceuticalresiduesinmunicipalwastewater,Environ.Int.45(2012)99– 111.
[5]K.Kümmerer,Significanceofantibioticsintheenvironment,J.Antimicrob. Chemother.52(2003)5–7.
[6]G.M.Bruce,R.C.Pleus,S.A.Snyder,ToxicologicalRelevanceofPharmaceuticals inDrinkingWater,Environ.Sci.Technol.44(2010)5619–5626.
[7]A.J.Watkinson,E.J.Murby, D.W.Kolpin,S.D.Costanzo,Theoccurrenceof antibioticsinanurbanwatershed:Fromwastewatertodrinkingwater,Sci. TotalEnviron.407(2009)2711–2723.
[8]M.Clara,N.Kreuzinger,B.Strenn,O.Gans,H.Kroiss,Thesolidsretentiontime— asuitabledesignparametertoevaluatethecapacityofwastewatertreatment plantstoremovemicropollutants,WaterRes.39(2005)97–106.
[9]S.Rodriguez-Mozaz,S.Chamorro,E.Marti,B.Huerta,M.Gros,A. Sànchez-Melsió,C.M.Borrego,D.Barceló,J.L.Balcázar,Occurrenceofantibioticsand antibioticresistancegenesinhospitalandurbanwastewatersandtheirimpact onthereceivingriver,WaterRes.69(2015)234–242.
[10]L. Kovalova, H. Siegrist, H. Singer, A. Wittmer, C.S. McArdell, Hospital WastewaterTreatmentbyMembraneBioreactor:Performanceand EfficiencyforOrganicMicropollutantElimination,Environ.Sci.Technol.46 (2012)1536–1545.
[11]I.Sires,E.Brillas,Remediationofwaterpollutioncausedbypharmaceutical residuesbasedonelectrochemicalseparationanddegradationtechnologies:A review,Environ.Int.40(2012)212–229.
[12]R.Tenne,K.Patel,K.Hashimoto,A.Fujishima,AnInternationalJournalDevoted toallAspectsofElectrodeKinetics,InterfacialStructure,Propertiesof Electrolytes,ColloidandBiologicalElectrochemistryEfficientelectrochemical reductionofnitratetoammoniausingconductivediamondfilmelectrodes,J. Electroanal.Chem.347(1993)409–415.
[13]M. Murugananthan, S. Yoshihara, T. Rakuma, N. Uehara, T. Shirakashi, Electrochemicaldegradationof17b-estradiol(E2)atboron-dopeddiamond (Si/BDD)thinfilmelectrode,ElectrochimicaActa.52(2007)3242–3249. [14]R.F. Brocenschi,R.C. Rocha-Filho,N.Bocchi,S.R. Biaggio,Electrochemical
degradationofestroneusingaboron-dopeddiamondanodeinafilter-press reactor,ElectrochimicaActa.197(2016)186–193.
[15]E.Brillas,I.Sirés,C.Arias,P.L.Cabot,F.Centellas,R.M.Rodríguez,J.A.Garrido, Mineralizationofparacetamolinaqueousmediumbyanodicoxidationwitha boron-dopeddiamondelectrode,Chemosphere.58(2005)399–406. [16]J.Boudreau,D.Bejan,S.Li,N.J.Bunce,CompetitionbetweenElectrochemical
AdvancedOxidationandElectrochemicalHypochlorinationof
SulfamethoxazoleataBoron-DopedDiamondAnode,Ind.Eng.Chem.Res.49 (2010)2537–2542.
[17]K.P.deAmorim,L.L.Romualdo,L.S.Andrade,Electrochemicaldegradationof sulfamethoxazoleandtrimethoprimatboron-dopeddiamondelectrode: Performance,kineticsandreactionpathway,Sep,Purif.Technol.120(2013) 319–327.
[18]M.Murugananthan,S.S.Latha,G.BhaskarRaju,S.Yoshihara,Roleofelectrolyte onanodicmineralizationofatenololatborondopeddiamondandPt electrodes,Sep.Purif.Technol.79(2011)56–62.
[19]G. Perez, A.R.Fernandez-Alba,A.M.Urtiaga,I. Ortiz,Electro-oxidation of reverseosmosisconcentratesgeneratedintertiarywatertreatment,Water Res.44(2010)2763–2772.
[20]A.M.Urtiaga,G.Pérez,R.Ibáñez,I.Ortiz,Removalofpharmaceuticalsfroma WWTPsecondaryeffluentbyultrafiltration/reverseosmosisfollowedby electrochemical oxidationoftheROconcentrate, Desalination.331(2013) 26–34. [21]Y.Lan,C.Coetsier,C.Causserand,K.G.Serrano,FeasibilityofMicropollutants TreatmentbyCouplingNanofiltrationandElectrochemicalOxidation:Caseof HospitalWastewater,Int.J.Chem.React.Eng.13(2015)153–159.
[22]K.Groenen-Serrano,WastewaterTreatmentbyElectrogenerationofStrong OxidantsUsingBorondopedDiamond(BDD),in:G.Kreysa,K.Ota,R.F.Savinell (Eds.),Encycl.Appl.Electrochem.,Springer,NewYork,2014,pp.2126–2132. [23]K.Serrano,P.A.Michaud,C.Comninellis,A.Savall,Electrochemicalpreparation ofperoxodisulfuricacidusingborondopeddiamondthinfilmelectrodes, ElectrochimicaActa.48(2002)431–436.
[24]I.M.Kolthoff, I.K.Miller,TheChemistry ofPersulfate.I.TheKineticsand MechanismoftheDecompositionofthePersulfateIoninAqueousMedium1,J. Am.Chem.Soc.73(1951)3055–3059.
[25]M.Murugananthan,S.S.Latha,G.BhaskarRaju,S.Yoshihara,Anodicoxidation ofketoprofen—Ananti-inflammatorydrugusingborondopeddiamondand platinumelectrodes,J.Hazard.Mater.180(2010)753–758.
[26]M.E.H.Bergmann,J.Rollin,T.Iourtchouk,Theoccurrenceofperchlorateduring drinkingwaterelectrolysisusingBDDanodes,ElectrochimicaActa.54(2009) 2102–2107.
[27]C.R.Costa,F.Montilla,E.Morallón,P.Olivi,Electrochemicaloxidationofacid black210dyeontheboron-dopeddiamondelectrodeinthepresenceof phosphateions:Effectofcurrentdensity,pH,andchlorideions,Electrochimica Acta.54(2009)7048–7055.
[28]V.Schmalz,T.Dittmar,D.Haaken,E.Worch,Electrochemicaldisinfectionof biologicallytreatedwastewaterfromsmalltreatmentsystemsbyusing boron-dopeddiamond(BDD)electrodes–Contributionfordirectreuseofdomestic wastewater,WaterRes.43(2009)5260–5266.
[29]O.Scialdone,S.Randazzo,A.Galia,G.Silvestri,Electrochemicaloxidationof organicsinwater:Roleofoperativeparametersintheabsenceandinthe presenceofNaCl,WaterRes.43(2009)2260–2272.
[30]Á.Anglada,A.Urtiaga,I.Ortiz,D.Mantzavinos,E.Diamadopoulos, Boron-dopeddiamondanodictreatmentoflandfillleachate:Evaluationofoperating variablesandformationofoxidationby-products,WaterRes.45(2011)828– 838.
[31]E.T. Urbansky,M.R.Schock, Issuesinmanaging therisksassociated with perchlorateindrinkingwater,J.Environ.Manage.56(1999)79–95. [32]B.P.Chaplin,Criticalreviewofelectrochemicaladvancedoxidationprocesses
forwatertreatmentapplications,Environ.Sci.Process.Impacts.16(2014) 1182–1203.
[33]K. Viswanathan, B.V. Tilak,Chemical,Electrochemical, andTechnological AspectsofSodiumChlorateManufacture,J,Electrochem.Soc. 131(1984)1551– 1559.
[34]M.S.Siddiqui,Chlorine-ozoneinteractions:Formationofchlorate,WaterRes. 30(1996)2160–2170.
[35]L.R.Czarnetzki,L.J.J.Janssen,Formationofhypochlorite,chlorateandoxygen duringNaClelectrolysisfromalkalinesolutionsatanRuO2/TiO2anode,J.Appl. Electrochem.22(1992)315–324.
[36]Y.J.Jung,K.W.Baek,B.S.Oh,J.-W.Kang,Aninvestigationoftheformationof chlorateandperchlorateduringelectrolysisusingPt/Tielectrodes:Theeffects ofpHandreactiveoxygenspeciesandtheresultsofkineticstudies,WaterRes. 44(2010)5345–5355.
[37]A.M. Polcaro, A. Vacca, M. Mascia, S. Palmas, J.R. Ruiz, Electrochemical treatmentofwaterswithBDDanodes:kineticsofthereactionsinvolving chlorides,J.Appl.Electrochem.39(2009)2083–2092.
[38]M.E.H.Bergmann,J.Rollin,Productandby-productformationinlaboratory studiesondisinfectionelectrolysisofwaterusingboron-dopeddiamond anodes,Catal.Today.124(2007)198–203.
[39]O. Azizi, D. Hubler, G. Schrader, J. Farrell, B.P. Chaplin, Mechanism of PerchlorateFormationonBoron-DopedDiamondFilmAnodes,Environ.Sci. Technol.45(2011)10582–10590.
[40]C. do, N. Brito, D.M. de Araújo, C.A. Martínez-Huitle, M.A. Rodrigo, Understandingactivechlorinespeciesproductionusingborondoped diamondfilmswithlowerandhighersp3/sp2ratio,Electrochem.Commun.55 (2015)34–38.
[41]S.Ferro,A.D.Battisti,I.Duo,C.Comninellis,W.Haenni,A.Perret,Chlorine EvolutionatHighlyBoron-DopedDiamondElectrodes,J.Electrochem.Soc.147 (2000)2614–2619,doi:http://dx.doi.org/10.1149/1.1393578.
[42]Y.Yang,J.J.Pignatello,J.Ma,W.A.Mitch,ComparisonofHalideImpactsonthe EfficiencyofContaminantDegradationbySulfateandHydroxylRadical-Based AdvancedOxidationProcesses(AOPs),Environ.Sci.Technol.48(2014)2344– 2351,doi:http://dx.doi.org/10.1021/es404118q.
[43]A.Donaghue,B.P.Chaplin,EffectofSelectOrganicCompoundsonPerchlorate FormationatBoron-dopedDiamondFilmAnodes,Environ.Sci.Technol.47 (2013)12391–12399,doi:http://dx.doi.org/10.1021/es4031672.
[44]I.Quesada,Y.Gonzalez,S.Schetrite,H.Budzinski,K.LeMenach,O.Lorain,N. Manier,S.AitAissa,P.Pandard,D.Abdelaziz,J.-M.Canonge,C.Albasi, PANACÉE:évaluationdufonctionnementd’unbioréacteuràmembranes immergéestraitantdeseffluentshospitaliersd’oncologie,Rev.Sci.Eau.28 (2015)1.
[45]ISO9562:2004(en)Waterquality—Determinationofadsorbableorganically boundhalogens(AOX)
[46]C.Racaud,A.Savall,P.Rondet,N.Bertrand,K.GroenenSerrano,Newelectrodes forsilver(II)electrogeneration:ComparisonbetweenTi/Pt,Nb/Pt,andNb/ BDD,Chem.Eng.J.211–212(2012)53–59.
[47] M.Panizza,P.A.Michaud,G.Cerisola,C.Comninellis,Anodicoxidationof 2-naphtholatboron-dopeddiamondelectrodes,J.Electroanal.Chem.507(2001) 206–214.
[48]C.Sonntag,U.vonGunten,ChemistryofOzoneinWaterandWastewater Treatment:FromBasicPrinciplestoApplications,IWAPublishing,2012. [49]H.Lutze, Sulfateradical based oxidation inwater treatment, Universität
Duisburg-Essen,2013.
[50]IonizationConstantsofInorganicMonoproticAcids,Chemistry.msu.edu. [51]P. Neta, V. Madhavan, H. Zemel, R.W. Fessenden, Rate constants and
mechanismofreactionofsulfateradicalanionwitharomaticcompounds,J. Am.Chem.Soc.99(1977)163–164.
[52]I.T.Osgerby, ISCO Technology Overview: Do You Really Understand the Chemistry?in:E.J.Calabrese,P.T.Kostecki,J.Dragun(Eds.),Contam.Soils SedimentsWater,Springer-Verlag,2006,pp.287–308.
[53]Y.Ji, C. Ferronato, A. Salvador, X. Yang, J.-M. Chovelon, Degradation of ciprofloxacinandsulfamethoxazolebyferrous-activatedpersulfate: Implicationsforremediationofgroundwatercontaminatedbyantibiotics,Sci. TotalEnviron.472(2014)800–808.
[54]M.M.Ahmed,S.Barbati,P.Doumenq,S.Chiron,Sulfateradicalanionoxidation ofdiclofenacandsulfamethoxazoleforwaterdecontamination,Chem.Eng.J. 197(2012)440–447.
[55]C.Liang,Z.-S.Wang,C.J.Bruell,InfluenceofpHonpersulfateoxidationofTCE atambienttemperatures,Chemosphere.66(2007)106–113.
[56]F. Minisci, A. Citterio, C. Giordano, Electron-transfer processes: peroxydisulfate,ausefulandversatilereagentinorganicchemistry,Acc. Chem.Res.16(1983)27–32.
[57] G.R.P.Malpass,D.W.Miwa,S.A.S.Machado,P.Olivi,A.J.Motheo,Oxidationof thepesticideatrazineatDSA1electrodes,J.Hazard.Mater.137(2006)565– 572.
[58]M.Panizza,G.Cerisola,Applicationofdiamondelectrodestoelectrochemical processes,ElectrochimicaActa.51(2005)191–199.
[59]T.N.Das,ReactivityandRoleofSO5"-RadicalinAqueousMediumChain OxidationofSulfitetoSulfateandAtmosphericSulfuricAcidGeneration,J. Phys.Chem.A.105(2001)9142–9155.
[60]H.Li,J.Ni,Electrogenerationofdisinfectionbyproductsataboron-doped diamondanodewithresorcinolasamodelsubstance,ElectrochimicaActa.69 (2012)268–274.
[61] C. Flox, E. Brillas, A. Savall, K. Groenen-Serrano, Kinetic Study of the ElectrochemicalMineralizationofm-CresolonaBoron-DopedDiamond Anode,Curr.Org.Chem.16(2012)1960–1966.
[62]R.Castagna,J.P.Eiserich,M.S.Budamagunta,P.Stipa,C.E.Cross,E.Proietti,J.C. Voss,L.Greci,Hydroxylradicalfromthereactionbetweenhypochloriteand hydrogenperoxide,Atmos.Environ.42(2008)6551–6554.
[63]A.Kraft,M.Stadelmann,M.Blaschke,D. Kreysig,B.Sandt,F.Schröder,J. Rennau,ElectrochemicalwaterdisinfectionPartI:Hypochloriteproduction fromverydilutechloridesolutions,J.Appl.Electrochem.29(1999)859–866.