Caractérisation des bases moléculaires de l'isolement reproducteur post-zygotique intrinsèque chez le Grand Corégone (Coregonus clupeaformis)
170
0
0
Texte intégral
(2) Caractérisation des bases moléculaires de l’isolement reproducteur post-zygotique intrinsèque chez le Grand Corégone (Coregonus clupeaformis) Thèse. Anne-Marie Dion-Côté. Sous la direction de :. Louis Bernatchez, directeur recherche.
(3) Résumé Alors que les technologies de séquençage à très haut débit ont permis de réaliser d’importants progrès pour documenter l’architecture génomique de la spéciation, les mécanismes moléculaires responsables de l’isolement reproducteur demeurent nébuleux. L’objectif principal de cette thèse est d’apporter un nouvel éclairage sur les bases moléculaires de l’isolement reproducteur dans un système d’espèces naissantes : le Grand Corégone. En particulier, l’approche était d’examiner les mécanismes associés à la stabilité du génome et à sa dérégulation dans un contexte de divergence et d’hybridation. Par une méthode de transcriptomique, nous avons documenté une profonde dérégulation à l’échelle du transcriptome chez les hybrides entre les formes naine et normale du Grand Corégone, incluant la dérépression des éléments transposables et des transcrits non-codants. Par la suite, nous avons observé que les hybrides montrent des signes importants de déstabilisation de la méiose et de la mitose, malgré l’absence de changement de caryotype entre les formes parentales. Finalement, nous avons observé un polymorphisme subchromosomique importants chez trois paires de Grand Corégones sympatriques, principalement associé à la période d’allopatrie. Ces travaux montrent que des changements importants à l’échelle du génome entier surviennent tôt au cours du processus de divergence, et sont susceptibles de conduire à de profonds dérèglements chez les hybrides. Enfin, cette thèse montre qu’une approche dite intégrative favorise une meilleure compréhension des mécanismes liés à la divergence et à la spéciation.. iii.
(4) Abstract While high throughput sequencing has led to significant progress towards the understanding of the genomic architecture of speciation, molecular mechanisms responsible for reproductive isolation remain unclear. The main objective of this thesis is to contribute to elucidate the molecular basis of reproductive isolation in a nascent species complex : the Lake Whitefish. In particular, the approach was to examine the mechanisms associated with genome stability and instability in a context of divergence and hybridization. Using transcriptomics, we documented a profound deregulation at the transcriptome scale in hybrids between dwarf and normal Lake Whitefish, including transposable element and non-coding RNA derepression. Then, we found that hybrids show significant signs of meiosis and mitosis breakdown, despite the absence of karyotype changes between parental forms. Finally, we observed conspicuous sub-chromosomal polymorphism in three sympatric Lake Whitefish species pairs, mainly associated with earlier allopatry. This work shows that genome-scale reorganization occurs early during divergence, and may lead to profound dysregulation in hybrids. This thesis also shows that integrative biology promotes a better understanding of the mechanisms linked to divergence and speciation.. iv.
(5) Table des matières RÉSUMÉ .......................................................................................................................................... III ABSTRACT ..................................................................................................................................... IV TABLE DES MATIÈRES ................................................................................................................V LISTE DES ABRÉVIATIONS......................................................................................................XII REMERCIEMENTS ...................................................................................................................... XV AVANT-PROPOS .......................................................................................................................XVII CHAPITRE 1 : INTRODUCTION GÉNÉRALE ...........................................................................1 1.1. LA SPÉCIATION COMME MÉCANISME GÉNÉRATEUR DE BIODIVERSITÉ ................................2. 1.2. BARRIÈRES À LA REPRODUCTION ............................................................................................2. 1.3. L’ISOLEMENT REPRODUCTEUR POST-ZYGOTIQUE INTRINSÈQUE RÉVÉLÉ PAR LES. HYBRIDES .............................................................................................................................................3. 1.4. CONTEXTE GÉOGRAPHIQUE DE LA SPÉCIATION .....................................................................4. 1.5. PROGRÈS RÉCENTS EN GÉNÉTIQUE DE LA SPÉCIATION .........................................................5. 1.6. MÉCANISMES DE L’ISOLEMENT REPRODUCTEUR POST-ZYGOTIQUE INTRINSÈQUE ............6. 1.6.1. MÉCANISMES GÉNÉTIQUES DE L’ISOLEMENT REPRODUCTEUR POST-ZYGOTIQUE. INTRINSÈQUE ........................................................................................................................................6. 1.6.2. MÉCANISMES TRANSCRIPTIONNELS DE L’ISOLEMENT REPRODUCTEUR POST-ZYGOTIQUE. INTRINSÈQUE ........................................................................................................................................7. 1.6.3. MÉCANISMES CHROMOSOMIQUES DE L’ISOLEMENT REPRODUCTEUR POST-ZYGOTIQUE. INTRINSÈQUE ........................................................................................................................................8. 1.7. LE RÔLE DE LA STABILITÉ DU GÉNOME EN SPÉCIATION ........................................................9. 1.7.1. LES ÉLÉMENTS TRANSPOSABLES ...........................................................................................10. 1.7.2. L’ANEUPLOÏDIE ......................................................................................................................10. 1.7.3. L’HÉTÉROCHROMATINE .........................................................................................................11. 1.8. LE GRAND CORÉGONE ...........................................................................................................12. 1.9. OBJECTIFS DE LA THÈSE .........................................................................................................14. v.
(6) CHAPITRE 2 : RNA-SEQ REVEALS TRANSCRIPTOMIC SHOCK INVOLVING TRANSPOSABLE ELEMENTS REACTIVATION IN HYBRIDS OF YOUNG LAKE WHITEFISH SPECIES ....................................................................................................................16 2.1. RÉSUMÉ ...................................................................................................................................17. 2.2. ABSTRACT................................................................................................................................18. 2.3. INTRODUCTION .......................................................................................................................19. 2.4. RESULTS ..................................................................................................................................22. 2.5. DISCUSSION .............................................................................................................................26. 2.6. MATERIAL AND METHODS ......................................................................................................35. 2.7. ACKNOWLEDGEMENTS ...........................................................................................................39. 2.8. TABLES.....................................................................................................................................40. 2.9. FIGURES ...................................................................................................................................43. 2.10. SUPPLEMENTARY METHODS ................................................................................................47. 2.11. SUPPLEMENTARY TABLE ......................................................................................................48. 2.12. SUPPLEMENTARY FIGURES ..................................................................................................49. CHAPITRE 3 : REPRODUCTIVE ISOLATION IN A NASCENT SPECIES PAIR IS ASSOCIATED WITH ANEUPLOIDY IN HYBRID OFFSPRING ...............................................59 3.1. RÉSUMÉ ...................................................................................................................................60. 3.2. ABSTRACT................................................................................................................................61. 3.3. INTRODUCTION .......................................................................................................................62. 3.4. MATERIAL AND METHODS .....................................................................................................65. 3.5. RESULTS ..................................................................................................................................68. 3.6. DISCUSSION .............................................................................................................................70. 3.7. DATA ACCESSIBILITY ..............................................................................................................77. 3.8. ACKNOWLEDGEMENTS ...........................................................................................................78. 3.9. TABLES.....................................................................................................................................79. 3.10. FIGURES.................................................................................................................................81. 3.11. SUPPLEMENTARY TABLES ....................................................................................................84. CHAPITRE 4 : CYTOGENETICS AND MISSED INFORMATION FROM GENOME SEQUENCING: STANDING CHROMOSOMAL VARIATION ASSOCIATED WITH REPRODUCTIVE ISOLATION IN LAKE WHITEFISH SPECIES PAIRS ...............................89 4.1. RÉSUMÉ ...................................................................................................................................90. vi.
(7) 4.2. ABSTRACT................................................................................................................................91. 4.3. INTRODUCTION .......................................................................................................................92. 4.4. MATERIAL AND METHODS ......................................................................................................95. 4.5. RESULTS ..................................................................................................................................99. 4.6. DISCUSSION ...........................................................................................................................103. 4.7. DATA ACCESSIBILITY ............................................................................................................109. 4.8. ACKNOWLEDGEMENTS .........................................................................................................110. 4.9. TABLES...................................................................................................................................111. 4.10. FIGURES...............................................................................................................................115. 4.11. SUPPLEMENTARY CODE .....................................................................................................121. 4.12. SUPPLEMENTARY TABLES ..................................................................................................125. 4.13. SUPPLEMENTARY FIGURES ................................................................................................131. CHAPITRE 5 : CONCLUSION ...................................................................................................133 5.1. RETOUR SUR LES PRINCIPAUX RÉSULTATS .........................................................................134. 5.2. PERSPECTIVES .......................................................................................................................137. 5.3. VERS UNE APPROCHE INTÉGRATIVE DE L’ÉTUDE DE LA SPÉCIATION ...............................139. CHAPITRE 6 : BIBLIOGRAPHIE ..............................................................................................140. vii.
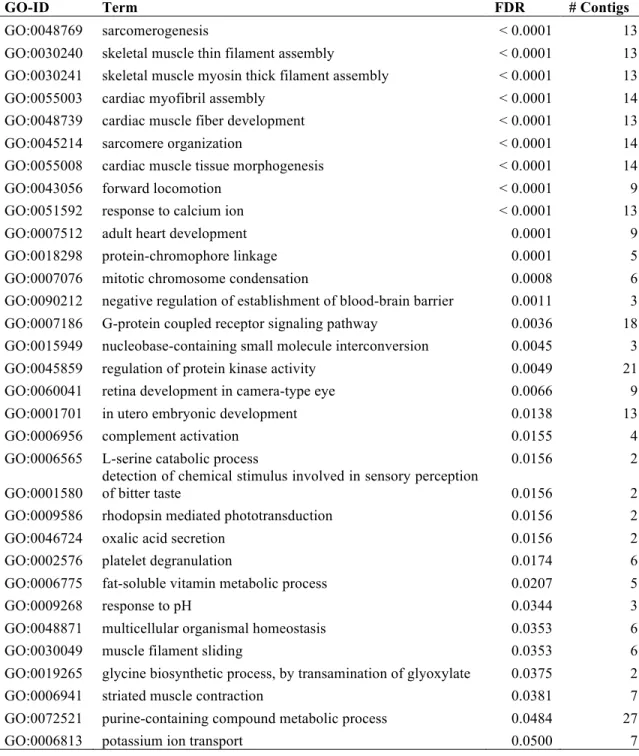
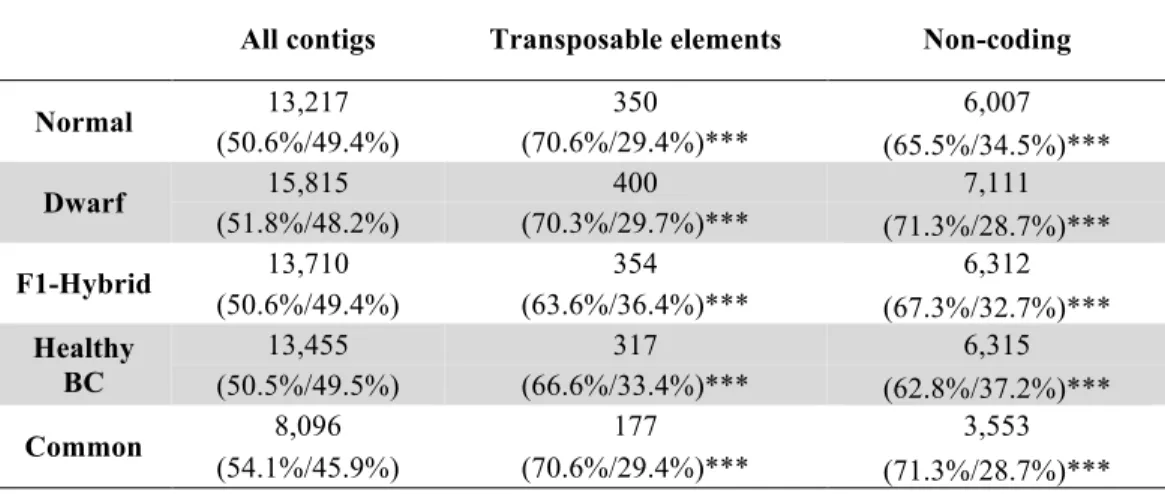
(8) Liste des tableaux TABLE 2.1. DIFFERENTIAL EXPRESSION SUMMARY. ........................................................... 40 TABLE 2.2. GO ENRICHMENT (BIOLOGICAL PROCESSES, FDR < 0.05) FOR DIFFERENTIALLY EXPRESSED TRANSCRIPTS BETWEEN PURE NORMAL AND DWARF WHITEFISH EMBRYOS (FDR < 0.01, FOLD-CHANGE > 2). .................................................... 41. TABLE 2.3. OVER-EXPRESSION OF TRANSPOSABLE ELEMENTS AND NON-CODING TRANSCRIPTS IN MALFORMED BACKCROSSES. .................................................................... 42. TABLE S 2.1. GO ENRICHMENT (BIOLOGICAL PROCESSES, FDR < 0.05) AMONG COMMONLY OVER-EXPRESSED TRANSCRIPTS IN MALFORMED BACKCROSSES AS COMPARED TO ALL OTHER GROUPS (FDR < 0.01, FOLD-CHANGE > 2). ............................. 48. TABLE 3.1. INDIVIDUALS SAMPLED IN THIS STUDY. ............................................................ 79 TABLE 3.2. SUMMARY STATISTICS OF CHROMOSOME NUMBER PER CROSS-TYPE AND GROUP. .................................................................................................................................. 80. TABLE S 3.1. SUMMARY STATISTICS FOR INDIVIDUAL FISH................................................ 84 TABLE S 3.2. ANOVA SUMMARY ON VARIATION COEFFICIENTS OF CHROMOSOME COUNTS PER INDIVIDUAL...................................................................................................... 86. TABLE S 3.3. TUKEY HSD POST-HOC TEST. ........................................................................ 87 TABLE S 3.4. FLIGNER-KILLEEN TEST (ON INDIVIDUAL MEDIAN CHROMOSOME COUNTS) .. ..................................................................................................................................... 88 TABLE 4.1. NUMBER OF INDIVIDUALS ANALYZED PER LAKE AND ECOTYPE WITH THEIR AVERAGE PHENOTYPIC CHARACTERISTICS. ...................................................................... 111. TABLE 4.2. FACTORS (SUPPLEMENTARY VARIABLES) WITH A SIGNIFICANT EFFECT ON DIMENSIONS 1 AND 3 FROM THE MULTIPLE FACTOR ANALYSIS. ....................................... 112. TABLE 4.3. SIGNIFICANT BARYCENTER POSITION ESTIMATES OF THE FACTOR LEVELS FOR WHICH MAIN FACTORS HAD A SIGNIFICANT EFFECT ON DIMENSIONS 1 AND 3 FROM THE MULTIPLE FACTOR ANALYSIS (TABLE 1). .......................................................................... 113. TABLE 4.4. CHROMOSOME MARKERS SIGNFICANTLY CORRELATED (P < 0.05) TO DIMENSIONS 1 AND 3 FROM THE MULTIPLE FACTOR ANALYSIS. ....................................... 114. TABLE S 4.1. INDIVIDUAL CHARACTERISTICS AND MARKERS SCORED. ........................... 125. viii.
(9) TABLE S 4.2. INDIVIDUAL CHARACTERISTICS AND IMPUTED MARKER VALUES USING THE IMPUTEMFA() FUNCTION. ................................................................................................. 128. ix.
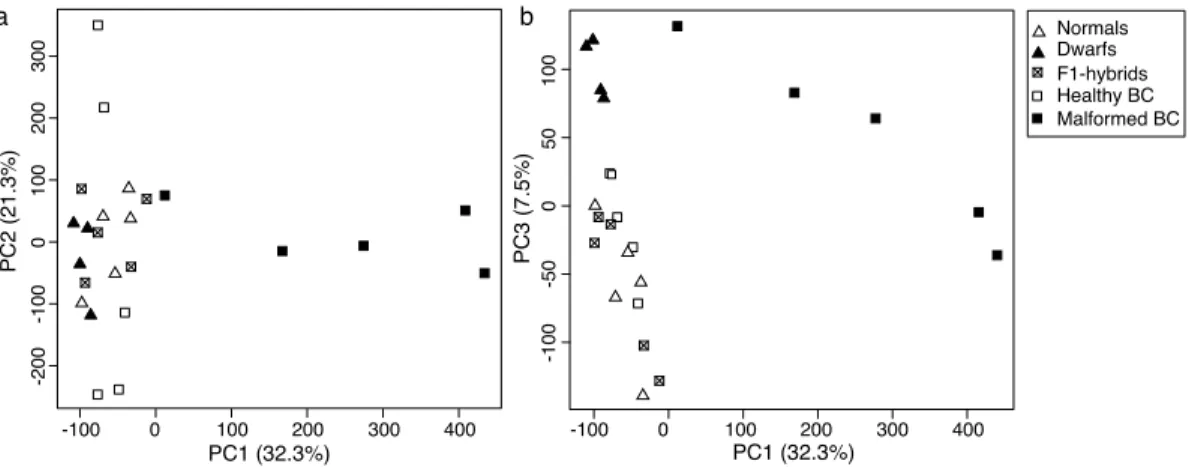
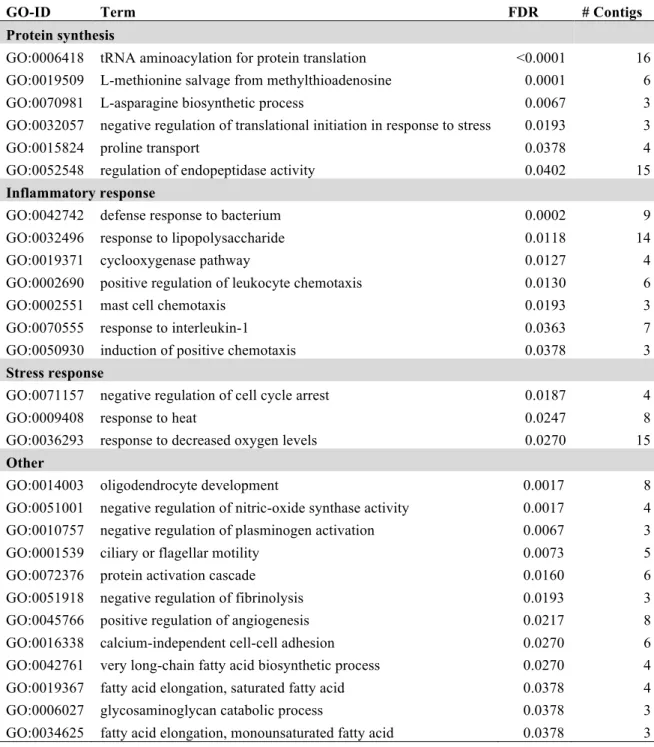
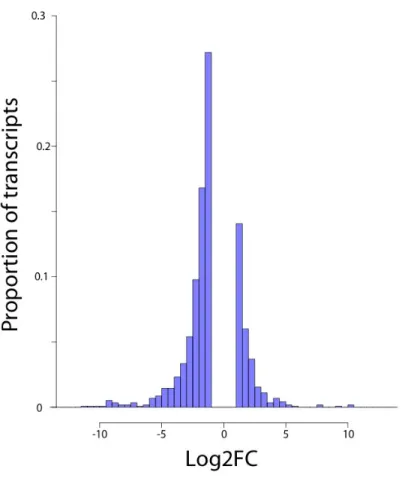
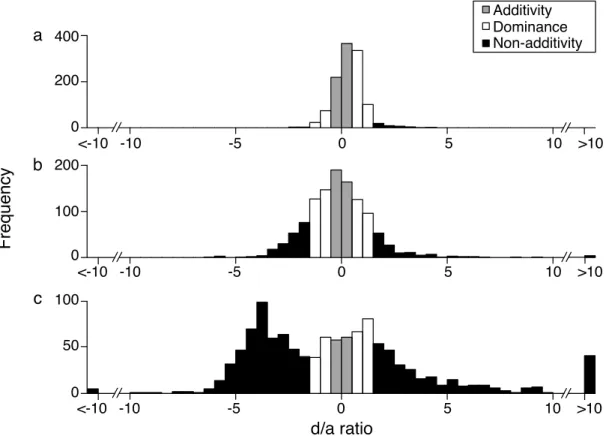
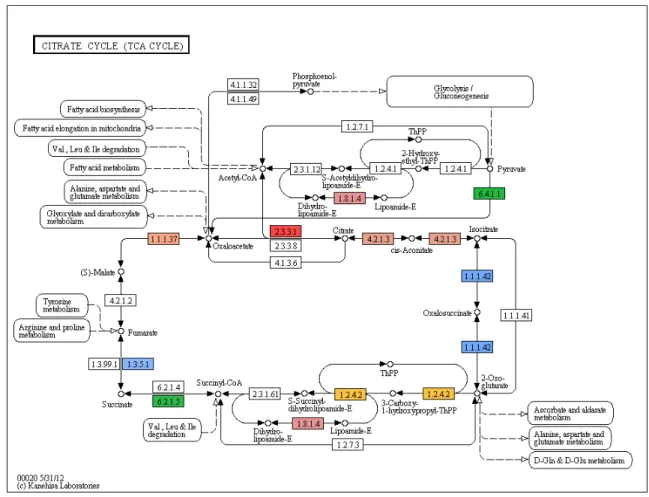
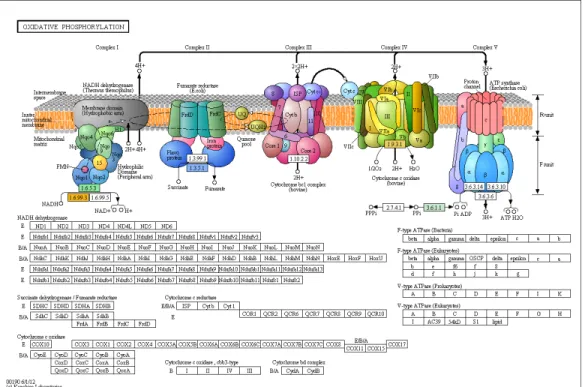
(10) Liste des Figures FIGURE 2.1. ANNOTATION SUMMARY OF THE ASSEMBLED TRANSCRIPTOME. ................... 43 FIGURE 2.2. TRANSPOSABLE ELEMENTS ANNOTATION. ...................................................... 44 FIGURE 2.3. UNIQUE TRANSCRIPTION PROFILE OBSERVED IN BACKCROSSES, AND MALFORMED BACKCROSSES IN PARTICULAR. ..................................................................... 45. FIGURE 2.4. OVER-EXPRESSED TRANSPOSABLE ELEMENT SUPER-FAMILIES IN MALFORMED BACKCROSSES IN ALL COMPARISONS (N = 125, FDR < 0.01, FOLD-CHANGE >. 2).. 46. FIGURE S 2.1. DISTRIBUTION OF VARIATION COEFFICIENT OF READ COUNTS FOR EACH CONTIG. ................................................................................................................................ 49. FIGURE S 2.2. DISTRIBUTION OF LOG2 FOLD-CHANGE (LOGFC) FOR TRANSCRIPTS DIFFERENTIALLY EXPRESSED BETWEEN DWARF AND NORMAL EMBRYOS (FDR < 0.01, FC. > 2). 50 FIGURE S 2.3. DISTRIBUTION OF LOG2 FOLD-CHANGE (LOG2FC) FOR TRANSCRIPTS DIFFERENTIALLY EXPRESSED IN MALFORMED BACKCROSS. .............................................. 51. FIGURE S 2.4. D/A RATIO DISTRIBUTION IN HYBRIDS. ......................................................... 52 FIGURE S 2.5. CARBOHYDRATE METABOLISM (PARTIAL) IS DOWN-REGULATED IN MALFORMED HYBRIDS.. ........................................................................................................ 53. FIGURE S 2.6. KEGG MAP OF THE TCA CYCLE. ................................................................ 54 FIGURE S 2.7. KEGG MAP OF THE OXIDATIVE PHOSPHORYLATION PATHWAY. ............... 55 FIGURE S 2.8. KEGG MAP OF FATTY ACID METABOLISM. ................................................. 56 FIGURE S 2.9. KEGG MAP OF THE PURINE METABOLISM. ................................................. 57 FIGURE S 2.10. KEGG MAP OF THE PYRIMIDINE METABOLISM. ....................................... 58 FIGURE 3.1. KARYOTYPES OF PURE PARENTAL FORMS AND ABNORMAL METAPHASES OF MALFORMED BACKCROSSES. ............................................................................................... 81. FIGURE 3.2. MEIOTIC BREAKDOWN IN MALFORMED BACKCROSSES REFLECTED BY THE ANALYSIS OF MITOTIC CHROMOSOMES. .............................................................................. 82. FIGURE 3.3. MITOTIC AND MEIOTIC CHROMOSOMAL INSTABILITY OCCURS IN BACKCROSSES BASED ON THE ANALYSIS OF MITOTIC CHROMOSOMES. ............................. 83. x.
(11) FIGURE 4.1. EXAMPLE OF CHROMATIN STRUCTURE POLYMORPHISM IN A NORMAL AND A DWARF INDIVIDUAL FROM EAST LAKE.. ............................................................................ 115. FIGURE 4.2. B CHROMOSOME IDENTIFIED IN LAKE WHITEFISH BY A) C-BANDING, B) CMA3/DAPI STAINING AND C) FISH WITH RDNA 28S (RED) AND RDNA 5S (GREEN) PROBES................................................................................................................................ 116. FIGURE 4.3. REPRESENTATIVE RDNA SITES POLYMORPHISM SHOWN BY FLUORESCENT IN SITU HYBRIDIZATION (FISH) WITH PROBES FOR 5S AND 28S RDNA. ............................ 117 FIGURE 4.4. PARTIAL CONSENSUS IDEOGRAM FOR ALL THREE SPECIES PAIRS SHOWING CHROMOSOME SHAPE AND ALL MARKERS IDENTIFIED ON CHROMOSOMES AND SCORED. ..... ................................................................................................................................... 118 FIGURE 4.5. MULTIPLE FACTOR ANALYSIS PERFORMED WITH FACTOMINER. .............. 119 FIGURE 4.6. MULTIPLE FACTOR ANALYSIS CORRELATION CIRCLE FOR DIMENSIONS 1 AND 3.. .................................................................................................................................. 120. FIGURE S 4.1. MULTIPLE FACTOR ANALYSIS PERFORMED WITH FACTOMINER. ........... 131 FIGURE S 4.2. MULTIPLE FACTOR ANALYSIS CORRELATION CIRCLE FOR DIMENSIONS 1 AND 3................................................................................................................................... 132. xi.
(12) Liste des abréviations ADN, ADNc, ADNg, ADNr. Acide désoxyribonucléique, ADN complémentaire, ADN génomique, ADN ribosomique. ANOVA. Analysis of variance (analyse de la variance). ARN, ARNm, miARN, ARNnc. Acide ribonucléique, ARN messager, micro ARN, ARN non-codant. BC. Backcross (rétro-croisement). BDM. Bateson-Dobzhansky-Muller. BLAST. Basic local alignment search tool (algorithme d’alignement de séquences). bp. Base pair (paire de bases). BP. Before present (avant ce jour). BWA. Burrows-Wheeler Aligner (outil d’alignement de séquences). cm. centimètre. DNMT. DNA methyltransferase (méthyltransférase d’ADN). EDTA. Ethylenediaminetetraacetic acid (acide éthylènediaminetétraacétique). eQTL. Expression quantitative trait locus (locus de trait quantitatif d’expression). FC. Fold-change (ratio de changement d’expression). FDR. False-discovery rate (taux de faux-positifs). FPKM. Fragments per kilobase of transcript per million reads (fragments par kilobase de transcrit par million de lectures alignées). g, kg, mg, µg, ng. Gramme, kilogramme, milligramme, microgramme, nanogramme. GO. Gene ontology (ontologie génique). MFA. Multiple factor analysis (analyse factorielle multiple). MMR. Mismatch repair (réparation des mésappariements). NF. Nombre fondamental. nr. Non-redundant database (base de données de séquences non-redondante). ORF. Open reading frame (cadre de lecture ouvert). xii.
(13) PCA, PC. Principal component analysis, principal component (analyse en composantes principales, composante principale). pH. Puissance hydrogène (mesure de l’acidité). RNA-seq. RNA-sequencing (séquençage d’ARN). RRBS. Reduced representation bisulfite sequencing (séquençage bisulfite à représentation réduite [du génome]). SNP. Single-nucleotide polymorphism (polymorphisme de nucléotide simple). TCA. Tricitric acid (acide tricritrique). U. Unité (activité d’enzyme). UV. Ultra-violet. YBP. Years before present (années avant ce jour). xiii.
(14) Sequencing of the human genome did not provide conclusive answers to old biological mysteries – instead, it showed that the right questions had not even begun to be asked. T Ryan Gregory (2005) The Evolution of the Genome. xiv.
(15) Remerciements J’aimerais d’abord remercier chaleureusement mon directeur Louis Bernatchez. Louis, tu m’as offert le plus beau cadeau qu’un mentor puisse offrir: la liberté d’explorer et de faire des découvertes inattendues, en plus des moyens pour y parvenir. Je te remercie également pour ton support, tant scientifique que moral. Tu as su tendre l’oreille aux moments opportuns, et me pousser lorsque j’en avais besoin. Mille fois merci! Je veux remercier les membres de mon comité d’encadrement: Christian Landry, Julie Turgeon et Nadia Aubin-Horth. Tous trois m’avez fait confiance à différentes étapes, et j’ai énormément apprécié les nombreux échanges scientifiques, professionnels et personnels que nous avons eus. Je vous dois une fière chandelle! Je voudrais également remercier mes plus proches collaborateurs, sans qui cette thèse n’existerait probablement pas. Sébastien Renaut, Eric Normandeau, Radka Symonová, Petr Ráb, Fabien Lamaze, Šárka Pelikánová: merci pour votre support, votre enthousiasme et votre audace. J’ai également eu l’immense plaisir de collaborer à d’autres études du laboratoire. Merci à Ciro Rico et Martin Laporte de m’avoir offert cette chance. Le laboratoire Bernatchez est une grande famille, où plusieurs gens nous épaulent. Dans le désordre, merci à Christopher Sauvage, Charles Perrier, Gregory Maes, Kim Praebel, Anne Dalziel, Jean-Sébastien Moore, Scott Pavey, Ben Sutherland et Bérénice Bougas, pour tout ce que vous avez fait pour moi. Merci aussi à Madoka Krick, Laura Benestan, Clément Rougeux, François-Olivier Hébert, Alysse Perreault-Payette, Simon Bernatchez, Élianne Valiquette et Geneviève Ouellet-Cauchon. Merci également à Guillaume Côté, Serge Higgins, Jean-Christopher Terrien, Jade Larivière, Lucie Papillon, Alain Goulet et Richard Janvier pour leur immense support technique. Merci à Thierry Gosselin, Vincent Bourret, Caroline Côté, Marie Filteau, Pierre-Alexandre Gagnaire, et Julie Jeukens pour leurs judicieux conseils.. xv.
(16) Lors de mes séjours en République Tchèque, j’ai eu l’immense plaisir de collaborer avec des gens extrêmement généreux. Merci à Zuzáná Majtanova, Petra Sejnohová et Alexandr Sember. Une pensée toute spéciale pour Jana Čechová. J’ai aussi le bonheur de remercier Karine Jacquet, une grande amie, et Jean-Yves Masson, mon superviseur de maîtrise. Merci pour vos questions, vos pistes de réponses, votre support et vos encouragements au long de mon doctorat. Merci également à mon amie Emilie, sans qui je ne serais possiblement jamais atterie là où je suis. Un merci tout spécial à mon « comité de relecture » : Jean-Sébastien Moore, Emilie Castonguay et Mathilde Dion-Côté. Je remercie également tous les organismes qui m’ont soutenue financièrement : le CRSNQ, Québec-Océan, le département de biologie de l’Université Laval, Québec-Océan, le FRQNT, la fondation Richard-Bernard, l’IBIS, la Société Provancher et l’AÉLIÉS. Enfin, aucun mot de suffirait à exprimer toute la gratitude que j’ai envers ma famille et mes amis pour leur support et leur patience au fil des ans, et en particulier envers Michel, mon père, Constance, ma mère, Charles-Olivier, Mathilde et, plus récemment, Marianne. Merci.. xvi.
(17) Avant-propos Cette thèse est organisée en 5 chapitres, incluant l’introduction générale (Chapitre 1) et la conclusion (Chapitre 5). Les chapitres 2, 3 et 4 sont publiés ou en voie de l’être dans des revues scientifiques. D’autre part, j’ai contribué à 2 autres articles publiés par des membres du laboratoire. Ceux-ci sont inclus en Annexe. Le chapitre 2 est publié sous la référence : Dion-Côté A-M, Renaut S, Normandeau E, Bernatchez L. 2014. RNA-seq reveals transcriptomic shock involving transposable elements reactivation in hybrids of young lake whitefish species. Molecular Biology and Evolution 31:1188–1199. AMDC et LB ont conçu le projet. LB a supervisé le projet. SR a effectué les croisements et récolté le matériel biologique. AMDC a produit les données. AMDC et EN ont analysé les données. AMDC a écrit le manuscrit en collaboration avec SR, EN et LB. Le chapitre 3 est publié sous la référence : Dion-Côté A-M, Symonová R, Ráb P, Bernatchez L. 2015. Reproductive isolation in a nascent species pair is associated with aneuploidy in hybrid offspring. Proceedings of the Royal Society B: Biological Sciences 282:20142862–20142862. AMDC et LB ont conçu le projet. LB et PR ont supervisé le projet. AMDC a acquis les données avec le support de SP. AMDC analysé les données en collaboration avec RS. AMDC a écrit le manuscrit en collaboration avec RS, PR et LB. Le chapitre 4 a été soumis à la revue Molecular Ecology : Dion-Côté A-M, Symonová R, Lamaze FC, Pelikánová S, Ráb P, Bernatchez L. Standing chromosomal variation associated with rapid divergence between nascent Lake Whitefish species. AMDC et LB ont conçu le projet. LB a supervisé le projet. AMDC a acquis les données avec le support de SP. AMDC, RS et FCL ont analysé les données. AMDC a écrit le manuscrit en collaboration avec RS, FCL, PR et LB.. xvii.
(18) Chapitre 1 : Introduction générale. 1.
(19) 1.1 La spéciation comme mécanisme générateur de biodiversité La spéciation est le processus évolutif par lequel est générée la biodiversité. On peut ainsi considérer l’espèce comme l’unité fondamentale de la biodiversité. Les mécanismes favorisant la spéciation, s’y opposant, et modulant sa vitesse, sont donc d’un intérêt central en biologie évolutive. Différents concepts d’espèces existent, ayant chacun leurs forces et leurs faiblesses selon le système d’étude (Coyne et Orr 2004). Dans le cadre de cette thèse, nous adopterons le concept biologique d’espèce puisque c’est celui qui capture le mieux les propriétés du système à l’étude. Ainsi, les espèces sont définies comme « des groupes de populations naturelles, effectivement ou potentiellement interfécondes, qui sont génétiquement isolées d’autres groupes similaires » (Mayr 1942). La spéciation est donc le processus évolutif par lequel les barrières à la reproduction se mettent progressivement en place, conduisant à l’apparition de lignées évolutives génétiquement divergentes et reproductivement isolées. Ces barrières peuvent être de natures écologique ou génétique, ou, plus vraisemblablement, une combinaison des deux, puisque toute entité biologique est déterminée par l’interaction de ces facteurs (Coyne et Orr 2004).. 1.2 Barrières à la reproduction L’implantation de barrières à la reproduction est essentielle au processus de divergence et de spéciation. En effet, en l’absence de barrières à la reproduction, des populations seront en mesure d’échanger librement des allèles (flux de gènes), prévenant la divergence (Slatkin 1987). L’isolement reproducteur peut se produire avant ou après la fertilisation : on dira qu’il est pré-zygotique ou post-zygotique. Les barrières pré-zygotiques réduisent la probabilité qu’il y ait fertilisation et favorisent ainsi la divergence en réduisant le flux de gènes entre populations. Citons à titre d’exemples le choix d’hôte chez les insectes phytophages et l’homogamie chez l’épinoche à trois épines (McKinnon et al. 2004; Nosil 2007). Les mécanismes d’isolement reproducteur post-zygotique peuvent être à leur tour classés comme extrinsèque, s’ils dépendent de l’environnement, ou intrinsèque, s’ils sont plutôt génétiques et indépendants de l’environnement. Bien qu’intuitive, cette classification n’est pas absolue. En effet, certains mécanismes peuvent présenter des caractéristiques de. 2.
(20) l’isolement reproducteur post-zygotique intrinsèque et extrinsèque. Par exemple, une éclosion asynchrone de larves hybrides repose vraisemblablement sur des mécanismes génétiques intrinsèques (Robison et al. 2001), mais sera également défavorable en raison du manque de synchronisme avec les ressources alimentaires d’un point de vue extrinsèque (Cushing 1990). Bien que les barrières pré-zygotiques semblent contribuer davantage à l’isolement reproducteur total (Coyne et Orr 1997), les barrières post-zygotiques sont les plus susceptibles d’être irréversibles (Muller 1939; Muller 1942). Cette thèse s’intéresse principalement aux mécanismes moléculaires de l’isolement post-zygotique intrinsèque.. 1.3 L’isolement reproducteur post-zygotique intrinsèque révélé par les hybrides Les hybrides offrent une fenêtre extraordinaire sur l’étude de la spéciation en raison de leurs propriétés uniques par rapport aux formes parentales (Maheshwari et Barbash 2011). En nature, les approches génomiques en zones hybrides ont montré que certaines portions du génome sont moins échangées entre lignées divergentes (ou introgressent moins), révélant ainsi que ces régions sont impliquées dans l’adaptation et l’isolement reproducteur (Abbott et al. 2013). En milieu contrôlé, les croisements hybrides permettent de mettre en évidence des interactions épistatiques complexes entre les génomes parentaux, notamment du point de vue transcriptionnel (Landry et al. 2007). Les croisements hybrides F2 (croisement de deux hybrides F1) ou rétro-croisés (croisement d’un hybride F1 avec l’une des forme parentales F0) présentent fréquemment un phénomène de rupture hybride (hybrid breakdown en anglais), alors que les hybrides de première génération (F1) peuvent ne montrer que peu ou pas de signes d’isolement reproducteur (e.g. Ellison et Burton 2008; Dey et al. 2014; Stelkens et al. 2015). Ce phénomène semble résulter d’interactions épistatiques (ou non-additives) entre allèles divergents des génomes parentaux (Rieseberg et al. 1999; Burton et al. 2006). De plus, les hybrides présentent fréquemment des phénomènes de dérégulation épigénétique, susceptibles d’induire à leur tour des changements d’expression génique et des aberrations de ségrégation chromosomique (Comai et al. 2003; Michalak 2009). Enfin, parce que les parents contribuent de façon inégale à la progéniture, par exemple via l’ADN mitochondrial transmis par la mère, les hybrides sont susceptibles de présenter des phénomènes d’incompatibilité et de. 3.
(21) débalancement uniques (Blier et al. 2001; Maheshwari et Barbash 2011; Burton et al. 2013). Ainsi, l’étude de l’hybridation permet de mettre en évidence les mécanismes impliqués dans l’isolement reproducteur post-zygotique intrinsèque entre lignées divergentes.. 1.4 Contexte géographique de la spéciation Historiquement, les barrières géographiques à la reproduction, en limitant le flux de gènes, étaient reconnues comme les plus importantes pour favoriser la spéciation (Mayr 1954a). Ainsi, la spéciation était étudiée dans son contexte géographique. Un système était donc caractérisé selon le niveau d’isolement géographique présent entre lignées divergentes. Cet isolement peut être complet (allopatrie), partiel (parapatrie) ou absent (sympatrie), ce qui influencera l’impact relatif des forces évolutives en jeu (Coyne et Orr 2004). En l’absence de flux de gènes, les populations isolées sont libres d’accumuler des différences génétiques par sélection divergente ou aléatoirement par dérive génétique (Haldane 1930; Coyne et Orr 2004). D’ailleurs, l’effet de la dérive et de la sélection sur le développement de l’isolement reproducteur entre populations isolées a pu être reproduit en laboratoire (Rice et Hostert 1993). En nature, la plus forte évidence supportant un rôle pour l’isolement géographique dans l’apparition de nouvelles espèces provient d’une concordance géographique entre espèces soeurs avec des barrières géographiques ou climatiques connues (Coyne et Orr 2004). Par exemple, la région des Grands Lacs en Amérique du Nord constitue une zone de suture, en raison de la glaciation du Pléistocène. Des populations de différentes espèces se sont retrouvées isolées dans différents refuges glaciaires, prévenant l’effet homogénéisateur du flux de gènes pendant plusieurs dizaines de milliers d’années, et favorisant ainsi l’apparition d’espèces soeurs (April et al. 2012). Au sens strict, la spéciation sympatrique se produit dans un contexte où les populations se chevauchent géographiquement et sont donc libres de se reproduire entre elles. Ainsi, la spéciation sympatrique de nature écologique requiert une source de sélection divergente, une forme d’isolement reproducteur, et un mécanisme génétique liant ces deux facteurs (Nosil 2012). Les exemples empiriques de spéciation purement sympatrique sont relativement rares, puisqu’il est souvent difficile de rejeter complètement l’hypothèse d’allopatrie. Ainsi, des exemples « classiques » de spéciation écologique sympatrique sont 4.
(22) en réalité plus complexes, par exemple par un rôle sous-estimé de la démographie ou une contribution de l’isolement géographique. En effet, un cas de spéciation sympatrique chez l’épinoche à trois épines semble plutôt résulter de deux vagues de colonisation successives des populations marines en eaux douces (Taylor et McPhail 2000). Dans le même ordre d’idées, l’exemple classique de divergence sympatrique chez l’insecte Rhagoletis pomonella aurait été favorisé par un flux de gènes entre populations originant du Mexique vers les populations états-uniennes (Feder et al. 2005). Enfin, le mode de divergence se situe généralement entre l’allopatrie stricte et la sympatrie pure, et ces deux situations sont plutôt les extrêmes d’un continuum de flux de gènes (Jiggins 2006). Une autre possibilité est la divergence dans un contexte mixte, c’est-à-dire d’une phase d’allopatrie suivie d’un contact secondaire (Nosil 2012). Les modes de spéciation mixtes sont susceptibles d’accélérer la fixation de certains réarrangements entre populations divergentes (Feder et al. 2011). Ainsi, les régions de contact secondaire constituent des zones particulièrement intéressantes pour l’étude de la divergence et de la spéciation, tel qu’évoqué précédemment pour le nord-est de l’Amérique du Nord.. 1.5 Progrès récents en génétique de la spéciation Un « gène de spéciation » peut être défini comme tout gène contribuant à l’évolution de l’isolement reproducteur entre populations divergentes (Nosil et Schluter 2011). On peut distinguer les bases génétiques de la spéciation liées à l’adaptation d’une part, et à l’isolement reproducteur d’autre part. Cette classification est imparfaite, puisque l’adaptation à des environnements divergents peut conduire à l’isolement reproducteur, mais elle permet de clarifier le point suivant. Avec le progrès des technologies de séquençage, la dernière décennie a été riche en découvertes dans le domaine de l’architecture génomique de la spéciation, et, en particulier, de l’adaptation (e.g. Colosimo et al. 2005; Hoekstra et al. 2006; Ellegren 2013; Renaut et al. 2013; Seehausen et al. 2014) Cependant, des progrès relativement timides ont été réalisés afin d’identifier les bases moléculaires directes de l’isolement reproducteur, et, plus particulièrement, des incompatibilités hybrides. Certes, il existe dans la littérature plusieurs exemples de « gènes de spéciation » impliqués directement dans l’isolement reproducteur (Presgraves 2010; Maheshwari et Barbash 2011; Nosil et Schluter 2011), mais, sauf exceptions (e.g. Ellison et. 5.
(23) Burton 2008; Gagnaire, Normandeau, et Bernatchez 2013), ces exemples ont été identifiés entre lignées relativement distantes (e.g. Russo et al. 1995; Maheshwari et Barbash 2011). Il demeure donc difficile d’évaluer si ces « gènes de spéciation » ont réellement contribué aux phases initiales du processus de divergence (Via et West 2008; Via 2009). Par ailleurs, les avancées réalisées grâce aux technologies de séquençage à très haut débit tendent à s’appuyer en grande partie sur les polymorphismes de nucléotide simple ou SNP (simple nucleotide polymorphism). Or, la caractérisation des meilleurs assemblages de génomes tels que chez l’humain a montré que les variations structurales et de nombre de copies sont plus fréquentes que les substitutions simples (Alkan et al. 2011). Malheureusement, plusieurs de ces variations telles que les séquences répétées, les insertions d’éléments transposables et les petites inversions demeurent extrêmement difficiles à caractériser avec les technologies actuelles (Treangen et Salzberg 2011; Ekblom et Wolf 2014). Ainsi, d’autres stratégies doivent être utilisées en complément des technologies de séquençage à haut débit afin de saisir l’ensemble des mécanismes génétiques impliqués en spéciation, et en particulier de ceux liés à l’isolement reproducteur.. 1.6 Mécanismes de l’isolement reproducteur post-zygotique intrinsèque L’isolement post-zygotique intrinsèque se traduit par des phénomènes non mutuellement exclusifs de stérilité et de non-viabilité des hybrides (Coyne et Orr 2004, p. 253). Il existe quatre grands mécanismes pouvant conduire à de telles incompatibilité hybrides : la présence d’endosymbiontes, les incompatibilités génétiques, différents niveaux de ploïdie et différents réarrangements chromosomiques (Coyne et Orr 2004). Les incompatibilités génétiques et les réarrangements chromosomiques sont les deux mécanismes abordés par cette thèse. 1.6.1. Mécanismes génétiques de l’isolement reproducteur post-zygotique intrinsèque. L’isolement post-zygotique intrinsèque conduit à la non-viabilité ou à la stérilité des hybrides. Or, comment est-il possible que la non-viabilité ou la stérilité hybride évoluent, alors qu’il s’agit de quelque chose de clairement contre-sélectionné (Orr et Presgraves 2000)? Conceptuellement, même Darwin reconnaissait qu’il est difficile pour une population de traverser une vallée adaptative pour arriver à un autre optimum adaptatif (Orr. 6.
(24) 1996). Ce paradoxe est résolu par le modèle de Dobzhansky-Muller (Dobzhansky 1937; Muller 1942). Ce modèle postule que deux populations isolées sont en mesure d’accumuler des mutations différentes à des loci épistatiques. Dans un contexte de contact secondaire ou d’hybridation, cette interaction peut être sous-optimale, conduisant à la stérilité ou à la mortalité des hybrides. Les hybrides de seconde génération (hybrides F2 ou rétro-croisés) et des générations suivantes (F3, etc.) tendent à montrer des manifestations plus prononcées des incompatibilités génétiques que les hybrides de première génération (ou F1). En effet, la recombinaison méiotique chez les hybrides F1 risque de briser des complexes de gènes co-adaptés. Le modèle de Dobzhansky-Muller a guidé de nombreuses études du domaine de la spéciation. On connaît aujourd’hui un certain nombres de gènes clairement impliqués dans des incompatibilités de type Dobzhansky-Muller, et ce, des invertébrés aux mammifères (Barbash et al. 2003; Presgraves et al. 2003; Ellison et Burton 2008; Mihola et al. 2009). 1.6.2. Mécanismes. transcriptionnels. de. l’isolement. reproducteur. post-zygotique. intrinsèque Les profils transcriptionnels peuvent être considérés comme des phénotypes intermédiaires entre les bases génétiques et le phénotype au sens commun (Aubin-Horth et Renn 2009). Les profils d’expression génique résultent de la co-évolution de dizaines de protéines (facteurs de transcriptions), de la séquence régulatrice des gènes et de modifications épigénétiques telles que les modifications des histones et la méthylation de l’ADN (Wray 2003). C’est dans les années 1970 que King et Wilson, s’appuyant sur l’exemple du chimpanzé et de l’humain, ont émis l’hypothèse voulant que les différences de régulation de l’expression des gènes pouvaient jouer un rôle clé en évolution et, en particulier, en spéciation (1975). De telles différences de régulation peuvent conduire chez les hybrides à des phénomènes importants de non-additivité, voire de transgressivité par rapport aux niveaux parentaux et ainsi être liées à l’isolement reproducteur (Wray 2003; Landry et al. 2007). Au-delà des interractions génétiques épistatiques, plusieurs études rapportent des phénomènes de dérégulation épigénétiques importants chez les hybrides, associés à une dérégulation de l’expression génique (Michalak 2009). Ainsi, l’étude des profils. 7.
(25) transcriptionnels entre lignées divergentes et leurs hybrides permet de mieux comprendre les mécanismes sous-jacents de l’isolement reproducteur post-zygotique intrinsèque. 1.6.3. Mécanismes. chromosomiques. de. l’isolement. reproducteur. post-zygotique. intrinsèque Le rôle des réarrangements chromosomiques en spéciation, principalement à savoir s’ils font partie des causes ou des conséquences de la divergence, demeure un sujet chaudement débattu en biologie évolutive (Faria et Navarro 2010). Il existe deux grands modèles théoriques concernant le rôle des réarrangements chromosomiques en spéciation. Les modèles classiques, et en particulier le modèle stasipatrique de White (1969; 1978a), reposent sur l’apparition de changements chromosomiques défavorables (sous-dominants) chez les hybrides (Faria et Navarro 2010). Ces réarrangements peuvent déstabiliser la méiose chez l’hétérozygote, à un point tel que les hybrides seront stériles (White 1978a; King 1993; Searle 1998). Or, il est peu probable qu’un réarrangement fortement sousdominant augmente en fréquence, puisque celui-ci devrait être rapidement éliminé par la sélection (Brown et O'Neill 2010). Ainsi, d’autres modèles ont dû être développés pour tenter de comprendre le rôle des réarrangements chromosomiques en spéciation. Rieseberg (2001) ainsi que Noor et al. (2001) ont proposé que les inversions contribuaient plutôt à limiter la recombinaison des régions impliquées, favorisant ainsi la divergence. Ces modèles, essentiellement de nature génique, postulent plutôt que les réarrangements chromosomiques, et particulièrement les inversions, favorisent des réductions locales du flux génique, se traduisant par une augmentation de la différenciation génétique à l’intérieur de ces réarrangements (Noor et al. 2001; Rieseberg 2001). Les réarrangements peuvent limiter la recombinaison chez l’hétérozygote par deux mécanismes : 1) en limitant les enjambements chromosomique (« crossovers ») ou 2) en conduisant à la production de gamètes non-balancés une fois recombinés (non-viables). Dans les deux cas, les régions à proximités du réarrangement tendent à moins recombiner chez les hybrides, et donc à diverger (Faria et Navarro 2010). Néanmoins, ces modèles ne concernent que les inversions, et négligent d’autres types de réarrangements chromosomiques tels que les fusion et fissions de chromosomes, les additions et délétions d’hétérochromatine, ou les translocations (King 1993). Le rôle des. 8.
(26) autres types de réarrangements chromosomiques dans l’isolement reproducteur est considéré comme neutre, sinon nébuleux (King 1993). Ainsi, il n’existe pas à l’heure actuelle de modèle complètement satisfaisant, et formulant des prédictions claires en tenant compte des divers phénomènes chromosomiques associés à la spéciation (Faria et Navarro 2010).. 1.7 Le rôle de la stabilité du génome en spéciation La stabilité du génome se définit comme un état où la séquence et la structure du génome demeurent constantes, et dans lequel le génome est en mesure d’assumer correctement ses fonctions, comme l’expression des gènes et la ségrégation fidèle des chromosomes. Par opposition, l’instabilité génomique est donc un état de dérégulation où les mutations surviennent à plus haute fréquence, que ce soit un changement de la séquence d’ADN, les réarrangements chromosomiques ou par l’aneuploïdie (Aguilera et Gómez-González 2008). Le concept de stabilité du génome est central en biologie moléculaire, mais peu utilisé en biologie évolutive. Or, il s’agit d’une propriété clée du génome, susceptible d’influencer de nombreux processus évolutifs, notamment par l’apport essentiel de la variation via la mutation au sein de la population (Lynch 2007). Plusieurs facteurs sont susceptibles de menacer la stabilité génomique, tant d’un point de vue intrinsèque qu’extrinsèque. Par exemple, le bris des points de contrôle du cycle cellulaire favorise la transmission d’erreurs de réparation ou réplication de l’ADN, consuisant ainsi à des mutations. Les agents mutagènes, comme les rayons UV, peuvent causer des cassures de l’ADN qui, lorsque non réparées, peuvent également conduire à des mutations ou des réarrangements chromosomiques (Aguilera et Gómez-González 2008). Par ailleurs, un certain nombre de gènes impliqués dans l’isolement reproducteur postzygotique intrinsèque est associé au maintien ou à la rupture de cette stabilité du génome (Presgraves 2010; Maheshwari et Barbash 2011). Une étude élégante réalisée chez la levure a montré en 2003 que la voie de réparation des mésappariements de l’ADN était responsable de la stérilité des hybrides entre souches divergentes, liant directement les mécanismes de stabilité du génome à l’isolement reproducteur (Greig et al. 2003). D’autres exemples illustrant l’importance de la stabilité du génome dans un contexte de divergence et de spéciation sont discutés ci-bas.. 9.
(27) 1.7.1. Les éléments transposables. Les éléments transposables sont des séquences d’ADN capables de se déplacer (« sauter ») à l’intérieur du génome de l’hôte via un intermédiaire d’ARN (Classe 1 : Rétrotransposons) ou via sa propre excision (Classe 2 : Transposons à ADN) (Feschotte et Pritham 2007; Levin et Moran 2011). Chez la majorité des métazoaires, seule une infime fraction du génome code pour des protéines (Lander et al. 2001; Mouse Genome Sequencing Consortium et al. 2002). Le reste du génome contient divers éléments structuraux (ex. centromères et télomères) et régulateurs (ex. promoteurs et miARNs), et bien sûr les éléments transposables. Les éléments transposables doivent impérativement être maintenus dans un état silencieux, sans quoi ils constituent une menace sérieuse pour la stabilité du génome, et conséquemment la valeur adaptative de l’individu (Levin et Moran 2011). Barbara McClintock a suggéré il y a plus de trente ans que l’hybridation puisse conduire à une dérépression des éléments transposables (McClintock 1984). Les conséquences de la dérépression des éléments transposables sont imprévisibles, mais peuvent aller de la dérégulation de la transcription, à l’interruption de cadres de lecture, et même au cancer (Feschotte 2008; Levin et Moran 2011). Plusieurs exemples de réactivation des éléments transposables résultant d’hybridation inter-spécifique sont rapportés dans la littérature, et sont parfois associés à la déméthylation de l’ADN (e.g. O'Neill et Graves 1998; Ungerer et al. 2006; Kelleher et al. 2012). Ainsi, l’hybridation est susceptible de conduire à des phénomènes d’instabilité génomique liée à la dérépression des éléments transposables. 1.7.2. L’aneuploïdie. L’aneuploïdie peut se définir comme étant la présence d’un nombre anormal de chromosomes, résultant d’erreurs lors de la ségrégation des chromosomes (Compton 2011; Gordon et al. 2012). Les mécanismes responsables de l’aneuploïdie sont nombreux et complexes. On retiendra que ces mécanismes peuvent conduire à la non-disjonction d’un ou plusieurs chromosomes en méiose ou en mitose (Compton 2011). En fonction du moment où cette non-disjonction se produit, l’individu sera entièrement aneuploïde ou présentera un mosaïcisme, c’est-à-dire une présence de cellules euploïdes et aneuploïdes au sein de l’organisme. Les conséquences de l’aneuploïdie sont variables, et dépendent de. 10.
(28) l’organisme à l’étude et du type d’aneuploïdie impliqué. De plus, la propension à produire des cellules aneuploïdes varie énormément d’une espèce à l’autre (de 1 cellule sur 1 million chez la levure à près de 1 sur 100 chez l’humain (Compton 2011)). Alors que la triploïdie est relativement bien tolérée chez les plantes (complément entier surnuméraire), la plupart des trisomies (un seul chromosome surnuméraire) sont non-viables chez l’humain (Siegel et Amon 2012). Les mécanismes moléculaires par lesquels l’aneuploïdie conduit à une réduction de la valeur adaptative pour l’individu demeurent incompris. Néanmoins, plusieurs auteurs s’accordent pour dire que le débalancement d’un chromosome conduit à un déséquilibre de la stoechiométrie des protéines (Torres et al. 2008; Compton 2011; Siegel et Amon 2012). De plus, il semble que l’aneuploïdie soit fortement associée à l’instabilité génomique, en plus de conduire à une augmentation des dommages à l’ADN (Sheltzer et al. 2011). Enfin, lorsque des chromosomes sur-numéraires sont présents, il semble que les machineries transcriptionnelle et de traduction puissent devenir limitantes, ce qui exerce une pression pour atteindre l’homéostasie cellulaire (Torres et al. 2007). À l’exception de quelques cas chez des espèces modèles comme la levure et la souris (Forejt et al. 2012; Hauffe et al. 2012; Charron et al. 2014), l’aneuploïdie n’a été que très rarement rapportée, voire étudiée, dans un contexte d’isolement reproducteur. 1.7.3. L’hétérochromatine. L’hétérochromatine est une forme de l’ADN nucléaire compactée grâce à des modifications de la molécule d’ADN elle-même (ex. méthylation des cytosines) et des histones autour desquelles l’ADN est enroulé (ex. hypo-acétylation et méthylation) (Grewal et Jia 2007). L’hétérochromatine joue un rôle clé dans nombre de processus nucléaires, et particulièrement dans le maintien de la stabilité du génome, notamment pour la régulation de la transcription, la recombinaison méiotique et la ségrégation des chromosomes (Eissenberg et Elgin 2014). Typiquement, l’hétérochromatine est constituée de séquences d’ADN hautement répétées telles que les satellites, les éléments transposables et les centromères. Pour cette raison, les séquences hétérochromatiniennes demeurent extrêmement difficiles à caratériser par les approches actuelles de séquençage et tendent à. 11.
(29) manquer même dans les assemblages de génomes les plus complets tel que celui de l’humain (Ekblom et Wolf 2014). L’hétérochromatine, et en particulier la méthylation de l’ADN, occupe un rôle central dans le maintien de la stabilité du génome (Weber et Schübeler 2007). L’ADN est méthylé tôt au cours du développement par une famille d’enzymes essentielles au développement nommées ADN méthyl-transférase (DNMT, DNA methyltransferase) (Jones 2012). La fonction de cette méthylation varie selon le contexte (site d’initiation de la transcription, corps du gène, centromère), mais on retiendra surtout son rôle de répresseur de la transcription et dans le maintien de l’hétérochromatine. La méthylation de l’ADN permet également de réprimer les éléments transposables. Enfin, la dérégulation de la méthylation de l’ADN a été associée à une augmentation de l’instabilité du génome, dont la présence de réarrangements chromosomiques et l’aneuploïdie (Grewal et Jia 2007).. 1.8 Le Grand Corégone Le Grand Corégone (Coregonus clupeaformis) est un poisson lacustre d’Amérique du Nord. Certaines populations sont caractérisées par l’occurrence en sympatrie d’une forme normale et d’une forme naine dérivée. Dans le bassin de la Rivière St-Jean dans le nord-est de l’Amérique du Nord, deux lignées glaciaires auraient été géographiquement isolées par l’avancée des glaciers il y a environ 60 000 ans, selon les plus récentes estimations basées sur le séquençage complet du génome mitochondrial (Jacobsen et al. 2012). Suite au retrait des glaciers (~12 000 ans), ces deux lignées, dites Atlantique et Acadienne, ont colonisé les lacs nouvellement formés, se retrouvant ainsi en sympatrie suite à un contact secondaire (Bernatchez et al. 2010). La compétition intra-spécifique pour les ressources (Landry et al. 2007), la disponibilité de niche écologique (Pigeon et al. 1997) et les pressions de sélection divergente (Lu et Bernatchez 1999) auraient ensemble favorisé la divergence de la forme naine limnétique depuis la forme normale benthique. Ces deux écotypes diffèrent par une gamme de traits phénotypiques, tels que la taille et le poids à l’âge adulte, l’âge de maturité sexuelle et le mode de vie. Alors que la forme normale benthique peut atteindre une taille de plus de 40 cm, un poids supérieur à 1 kg et la maturité à 3-4 ans, la forme naine limnétique n’atteint qu’une taille d’environ 20 cm pour un poids de 100g, et mature dès 1-2 ans (Bernatchez et al. 2010). Des études transcriptomiques réalisées à partir de micro-puces. 12.
(30) à ADN ont également montré des différences d’expression génique importantes entre les formes naine et normale (Derome et al. 2006; St-Cyr et al. 2008; Whiteley et al. 2008; Jeukens et Bernatchez 2011). Dans un contexte de divergence sympatrique, c’est l’implantation de barrières à la reproduction efficaces qui permet de consolider un tel niveau de divergence. Il existe des preuves directes et indirectes de l’isolement pré-zygotique chez les corégones. De par leur différence de taille, les corégones nains et normaux sont davantage suceptibles de se reproduire avec leurs semblables (McKinnon et al. 2004). De plus, des données non publiées suggèrent que les formes naines et normales se reproduisent à des moments et lieux différents dans certains plans d’eau. Par ailleurs, puisqu’il est possible de réaliser des croisements contrôlés entre les formes naines et normales du Grand Corégone, nous savons qu’il existe des barrières post-zygotiques extrinsèques et intrinsèques puissantes entre les formes naine et normale. La mortalité embryonnaire des croisements hybrides F1 et rétrocroisés est beaucoup plus importante que celle des croisements purs nains et normaux (~50% et ~70% respectivement, contre ~20%; Lu et Bernatchez 1998; Rogers et Bernatchez 2006). Parmi les embryons rétro-croisés, une proportion importante se développe anormalement (10-30%) et montre un phénotype malformé (Renaut et Bernatchez 2011; Dion-Côté et al. 2015). De plus, l’émergence des larves rétro-croisées est asynchrone comparativement aux larves pures, ce qui peut conduire à un découplage avec les ressources trophiques disponibles (Rogers et Bernatchez 2006). Enfin, des études de transcriptomique ont montré des phénomènes de dérégulation transcriptionnelle importante chez ces hybrides rétro-croisés malformés, associés à une sous-régulation des gènes essentiels au développement (Renaut et Bernatchez 2011). Ainsi, le Grand Corégone est un excellent système pour étudier les bases moléculaires de l’isolement reproducteur. D’une part, des études ont montré que les différences phénotypiques observées sont en bonne partie déterminées génétiquement (Bernatchez et al. 2010). D’autre part, puisque ces formes ont divergé récemment à l’échelle évolutive (~12 000 ans), il est plus probable que les incompatibilités identifiées aient réellement contribué au processus de divergence, plutôt que de s’être accumulées par la suite (Via et West 2008; Via 2009). Enfin, la possibilité de réaliser des croisements hybrides en laboratoire permet d’étudier directement les barrières à la reproduction chez les hybrides.. 13.
(31) 1.9 Objectifs de la thèse L’objectif général de ma thèse était d’apporter un nouvel éclairage sur les bases moléculaires de l’isolement post-zygotique dans un système d’espèces naissantes. Mon approche cible les causes mécanistiques de l’isolement reproducteur, par opposition à la recherche de gènes de spéciation. Plus spécifiquement, j’investigue l’importance de la stabilité du génome et de sa réorganisation tôt dans le processus de spéciation. Mes travaux apportent des éléments de réponses à deux questions d’actualité dans le domaine de l’étude de la spéciation (Marie Curie SPECIATION Network 2012). Premièrement, quelles sont les barrières contribuant à l’isolement reproducteur au début du processus de spéciation? Ensuite, quelles sont les conditions environnementales et génétiques favorables à la divergence et, ultimement, à la spéciation? Dans le second chapitre, nous avons voulu tester directement l’hypothèse de la dérépression des éléments transposables chez les hybrides. En effet, des travaux réalisés au laboratoire suggéraient que les éléments transposables puissent être déréprimés chez les hybrides du Grand Corégone (Renaut et Bernatchez 2011). Par une approche de séquençage d’ARN, nous avons confirmé que les éléments transposables et les ARNs non-codants étaient déréprimés chez les hybrides rétro-croisés malformés. Cette étude a également permis de confirmer les phénomènes de transgressivité transcriptionnelle chez les hybrides et de découvrir de nouveaux gènes candidatsde la divergence adaptative entre les Corégones nains et les normaux. Le troisième chapitre visait à tester l’hypothèse que l’instabilité génomique contribue à l’isolement reproducteur dans le système du Grand Corégone. Pour ce faire, nous avons directement examiné les chromosomes d’hybrides rétro-croisés sains et malformés, ce qui nous a permis de mettre en évidence un phénomène d’aneuploïdie généralisée. Plus précisément, nous avons découvert que les comptes de chromosomes variaient beaucoup d’une cellule à l’autre chez les individus rétro-croisés sains, une observation cohérente avec une instabilité mitotique de la ségrégation des chromosomes. D’autre part, nous avons découvert des individus aux comptes chromosomiques très variables chez les rétro-croisés malformés, certains étant haploïdes, diploïde, triploïde et même presque tétraploïde. Cette observation suggère fortement une instabilité méiotique prononcée chez leur parent hybride. 14.
(32) F1, conduisant vraisemblablement à la non-disjonction du complément chromosomique entier. Dans le quatrième et dernier chapitre, nous avons appliqué des techniques de cytogénétique classique et moléculaire afin de vérifier la présence de réarrangements chromosomiques au sein de trois paires d’espèces naissantes du Grand Corégone. Cette approche nous a permis de mettre en évidence un polymorphisme sub-chromosomique inattendu, reflétant vraisemblablement un processus de réorganisation génomique en cours. Nous avons également appliqué une stratégie inédite de statistiques multi-variées aux données cytogénétiques qui nous a permis de détecter des patrons de divergence chromosomiques entre lignées glaciaires, entre lacs, et entre écotypes.. 15.
(33) Chapitre 2 : RNA-seq reveals transcriptomic shock involving transposable elements reactivation in hybrids of young lake whitefish species. 16.
(34) 2.1 Résumé L’identification des bases moléculaires de l’isolement reproducteur entre lignées divergentes est une étape essentielle vers la compréhension de la spéciation dans les populations naturelles. Les barrières à la reproduction peuvent conduire à la rupture hybride, un syndrome documenté dans plusieurs systèmes et impliquant potentiellement la réactivation des éléments transposables. Dans l’Est de l’Amérique du Nord, deux lignées du Grand Corégone ont colonisé plusieurs lacs postglaciaires il y a ~12 000 ans, et une espèce naine limnétique a évolué de façon répétée à partir de l’espèce normale benthique. L’isolement reproducteur est incomplet entre ces formes : des hybrides viables peuvent être générés en laboratoire, mais une mortalité significative chez les hybrides rétro-croisés est observée, associée à un phénotype malformé, suggérant un phénomène de rupture hybride. Par des analyses de séquençage d’ARN, l’objectif de cette étude était d’identifier les gènes mal exprimés chez ces hybrides et de rigoureusement tester l’hypothèse de réactivation des éléments transposables chez ceux-ci. Nous avons comparé le profil transcriptionnel d’embryons purs, hybrides F1, et d’hybrides rétro-croisés sains et malformés à un stade embryonnaire tardif. Pour la première fois, des différences d’expression prononcées, cohérentes avec la divergence adaptative documentée par des études antérieures chez les adultes, ont été identifiées entre les embryons purs. Une profonde dérégulation à l’échelle du transcriptome a été observée chez les rétro-croisés malformés, avec plus de 15% des transcrits différentiellement exprimés pour toutes les comparaisons, comparativement à 1,5% entre les formes parentales. De solides évidences supportant l’hypothèse de la réactivation des éléments transposables et des transcrits non-codants sont présentées. Nous suggérons que la rupture hybride résulte probablement de nombreuses incompatibilités génomiques, incluant vraisemblablement les éléments transposables. Combinés aux études antérieurest, ces résultats révèlent une synergie entre plusieurs barrières à la reproduction, contribuant au maintien de la divergence entre ces deux jeunes espèces de corégones.. 17.
(35) 2.2 Abstract Identifying the molecular basis of reproductive isolation among diverging lineages represents an essential step toward understanding speciation in natural populations. Postzygotic barriers can lead to hybrid breakdown, a syndrome that has been documented in several systems, potentially involving the reactivation of transposable elements. In northeastern North America, two lake whitefish lineages have repeatedly colonized postglacial lakes ~12,000 years ago, and a dwarf limnetic species has evolved multiple times from the normal benthic species. Reproductive isolation is incomplete between them; viable hybrids can be generated in the laboratory but significant mortality occurs and is associated with a malformed phenotype in backcross embryos, thus revealing a hybrid breakdown syndrome. By means of RNA-seq analyses, the objective of this study was to determine which genes were misregulated in hybrids and rigorously test the hypothesis of transposable element reactivation. We compared the transcriptomic landscape in pure embryos, F1-hybrids, and healthy and malformed backcrosses at the late embryonic stage. Extensive expression differences consistent with previously documented adaptive divergence between pure normal and dwarf embryos were identified for the first time. Pronounced transcriptome-wide deregulation in malformed backcrosses was observed, with over 15% of transcripts differentially expressed in all comparisons, compared with 1.5% between pure parental forms. Convincing evidence of transposable elements and noncoding transcripts reactivation in malformed backcrosses is presented. We propose that hybrid breakdown likely results from extensive genomic incompatibilities, plausibly encompassing transposable elements. Combined with previous studies, these results reveal synergy among many reproductive barriers, thus maintaining divergence between these two young whitefish species.. 18.
Figure
+7

Documents relatifs
Les résultats obtenus de la comparaison des moyennes de la teneur en lipides dans la chair des crevettes entre les mâles et les femelles d'une même espèce ne
Ces espaces ont été introduits par Hilbert en 1909, pour développer l’Analyse fonc- tionnelle abstraite, dont le point de départ consiste à considérer des fonctions comme
Mais si ce terme est aussi maintenant présent dans la langue générale et la presse (ce qui est une fois encore révélateur de la manière dont ces applications nous sont de
Pour notre enquête, il faut donc aller plus loin et regarder en premier lieu les textes rabbiniques, où Alexandre apparaît bien plus de fois 2 : onze épisodes lui
L’origine de ces différences de résultats n’est pas connue. Une hypothèse est que l’expression de ce gène serait plus sensible à une réduction de la
Pour tester le rôle de SH dans le spectre d’hôte, la seconde partie de ma thèse a consisté à développer un système de génétique inverse pour le Fr-AMPV-C permettant la
De même que l’État islamique avait été assimilé à un artefact occidental (CIA, Mossad…), certaines théories circulant sur les réseaux sociaux ont interprété
attributions were associated with partners’ lower relationship satisfaction, and global and stable attributions were related to lower partner sexual satisfaction. Partner’s
