Contribution à l’étude des peptides obliques impliqués dans des transconformations
Texte intégral
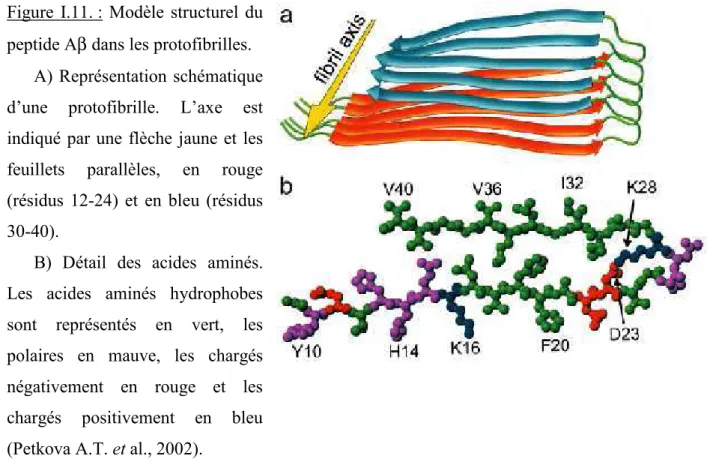
Figure









Documents relatifs
On dit que la courbe C admet la droite pour asymptote oblique en + ∞ (on précise « oblique » car le coefficient directeur de est non nul). 3°) On admet que
Et puisqu’on en est aux mutations de ce temps, je viens de lire que dans mon hôpital on se propose de transformer le service de pédiatrie en géria trie, tandis que
Nouvelle lampe électrique à rhéophores circulaires obliques..
Le système nerveux central (SNC) joue un rôle clé dans la détection et le contrôle du statut énergétique de l'organisme (Meyers J, et al., 2012) et l'hypothalamus en particulier
Le pp produit par les cellules pp ou cellules F qui représentent le quatrième type de cellules endocrines pancréatiques, elles sont principalement situées à la
Soit S le sommet d'une cône circulaire oblique coupé par un plan X sui- vant la courbe AMA^N; je cherche la ligne droite suivant laquelle l'un des plans cycliques coupe le plan X et
Appelons podaire oblique d'une courbe, par rapport à un point O, le lieu des pieds, sur les tangentes à cette courbe, des droites partant du point O et faisant avec les tangentes
De l'expression finale (4) résulte bien l'expression annoncée de la somme des aires, puisque chacun des triangles T ou T' a pour
