CO2 Removal from a CO2–CH4 Gas Mixture by Clathrate Hydrate Formation Using THF and SDS as Water-Soluble Hydrate Promoters
13
0
0
Texte intégral
Figure
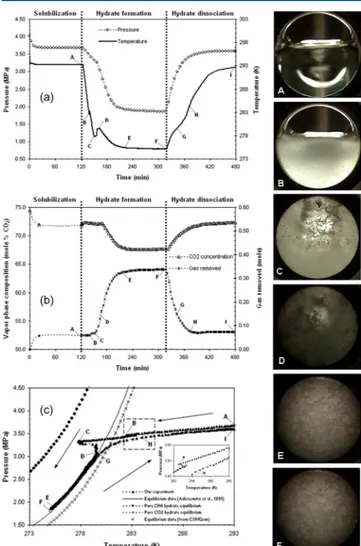


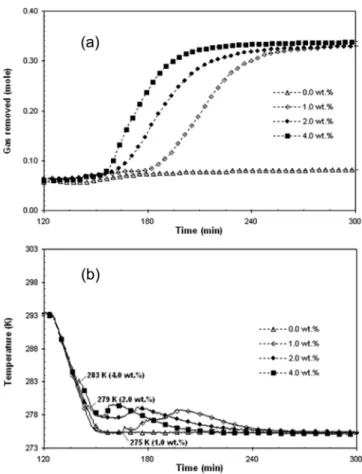
+4



Documents relatifs