METHODOLOGIES FOR THE REGIOCONTROLLED
ASSEMBLY OF α-SUBSTITUTED BUTENOLIDES AND
5-HYDROXYPYRROL-2(5H)-ONES;
ENANTIOSELECTIVE TOTAL SYNTHESIS OF
ANNOMOLON A & AUXOFURAN
Thèse
Richard Loach
Doctorat en chimie
Philosophiae Doctor (Ph.D.)
Québec, Canada
© Richard Loach, 2013
iii
RÉSUMÉ
Les travaux de cette thèse concernent des synthèses totales de produits naturels d’importance biologique, qui ont pu être réalisées grâce au développement de méthodologies pour synthétiser des hétérocycles oxygénés et azotés.
Le premier chapitre comprend le développement d’une méthodologie pour synthétiser des buténolides substitués en alpha, en utilisant le 3-bromo-2-silyloxyfurane pour former un nucléophile très général via échange lithium-halogène, et a démontré une excellente réactivité avec une grande variété d’électrophiles. La flexibilité de ce protocole nous a permis de compléter la première synthèse totale de l’annomolon A, une acétogénine ayant des propriétés anti-tumorales prometteuses. Notre synthèse a été réalisée de manière hautement convergente en combinant notre méthodologie pour α-alkylation/γ-oxyfonctionnalisation avec le couplage de métathèse croisée afin d’unir les deux segments clés de la molécule. Celles-ci contribuent à un rendement global de 18% pour les 17 étapes de notre synthèse, ce qui représente non seulement la première d’une hydroxybuténolide acétogénine, mais offre également une stratégie globale pour atteindre les autres membres de cette nouvelle série de composés naturels.
En parallèle avec ce projet, nous avons réussi à agrandir la chimie d’oxyfonctionnalisation, laquelle emploie le diméthyldioxirane comme oxydant très doux, pour la synthèse des 5-hydroxypyrrol-2(5H)-ones. La procédure, caractérisée par un haut niveau de régio et chimiosélectivité, tolère une grande variété de substituants sur les précurseurs, qui sont accédés rapidement à partir d’acides furoїques.
Le deuxième chapitre concerne la synthèse énantiosélective de l’auxofuran, un produit naturel avec des applications potentielles dans le domaine d’agrochimie. La structure de cette tétrahydrobenzofurane est très originale, et notre synthèse est non seulement la première mais elle permet la confirmation de la structure proposée pour l’auxofuran, ainsi que sa
iv
configuration absolue. Notre voie consiste d’une réaction Diels Alder-rétro-Diels Alder entre une oxazole et un acétylène, ce qui offre aussi la possibilité de synthétiser des analogues. L’exploration de cette chimie nous a permis de réaliser une méthodologie plus générale et très puissante pour construire des furanes 3,4-disubstitués à partir des acétylènes non-activés. Ainsi, son utilité a été bien démontrée par les synthèses d’anhydrides maléїques bioactives, y compris l’anhydride chaetomellique A, et les membres d’anhydrides et imides de camphorata.
v
REMERCIEMENTS/ACKNOWLEDGEMENTS
This work would not have been possible without the constant support and inspiration of my research supervisor Professor John Boukouvalas. John, thank you for having given me the opportunity to initiate and see through so many exciting projects. It was a highly rewarding experience to have been able to contribute to the stimulating research in your laboratory, and a real honour to have learnt how to do chemistry under your tutelage. Your peerless wit and talent for writing can only be matched by your knowledge and wisdom, which have at times seemed infinite. Your infectious enthusiasm and passion for organic chemistry and the art of total synthesis kept me primed with the sort of energy that allowed me to persist and persevere in the most challenging of moments (and weather!).
I would also like to take this opportunity to express my heartfelt thanks to the wider community of colleagues and friends in the chemistry department at Laval that I have been lucky enough to encounter during my time in Québec. Paola, fue un placer ser initiado al laboratorio bajo tu supervision, tu me diste esa confianza al comienzo cuando mas lo necesitaba. Wang, I am grateful for the six months I was fortunate enough to work next to you and learn so much. Lucas, it was always a jolly good laugh in the lab with you around; Nicolas, merci pour les cours de “québécois” – je vous souhaite beaucoup de chance pour l’avenir. Charles, c’était un plaisir d’apprendre de ton perfectionnisme et diligence, et Vince, it has been fantastic to observe how efficiently you work, always on the right track and always managing to get the product no matter what. Raphaël, je ne pense pas avoir rencontré quelqu’un possédant autant de joie de vivre que toi, ton énergie positive et attitude envers les problèmes est exemplaire. Je tiens à remercier aussi les autres collègues qui m’ont aidé et encouragé durant mes études, surtout Baptiste, Pierre Morin, Israel (thanks for the football matches mate!), Marc-André et Gauthier. I would also like to thank Professor Peter McBreen for having allowed me to participate in the exciting collaborations between our two research groups.
vi
I wish to also express my immense gratitude towards my family for their unstinting support and fervent belief in my abilities, especially my parents Peter and Alicia for instilling in me the values of hard work, curiosity and humility. Je veux aussi remercier ma famille de ce côté de l’Atlantique qui m’a appuyé tout le long de mes études – surtout Denis et Francine. Thanks too to Jules, a fantastic and loyal friend who was always there to help in times of need.
Finally, and most importantly, thank you a million times over to my wife, Manon, and daughters Eva and Liliana, for whom all the pages in this thesis would not have enough space to express my appreciation and awe at having them in my life. Thank you for keeping my feet on the ground, for keeping me standing up and keeping me smiling. I hope I can do you proud.
vii
TABLE OF CONTENTS
Résumé... iii
Remerciements... v
Table of Contents... vii
List of Tables... xiii
List of Figures... xv
List of Schemes... xvii
Abbreviations... xix
Chapter 1: General Introduction
...1
1.1) Introduction to the Total Synthesis of Natural Products
...3
1.1.1) The beginnings of natural product synthesis... 3
1.1.2) Maturing of a discipline: the Woodwardian era... 5
1.2) The Utility of Total Synthesis
...9
1.2.1) Structural determination and new chemistry development... 9
1.2.2) Biological importance... 10
1.3) Modern Total Synthesis
...13
1.3.1) The holy trinity: chemo-, regio- and stereoselectivity... 13
1.3.2) Cross-coupling... 16
1.3.3) Working towards the ideal, greener synthesis... 18
1.3.3.1) The concept of “ideality”... 18
1.3.3.2) Green chemistry and total synthesis... 22
1.3.3.3) Click chemistry... 25
1.3.4) Thesis objectives... 27
1.4) References
...28
RESULTS AND DISCUSSION
Chapter 2: Methodologies for the Regiocontrolled Assembly
of α-Butenolides and 5-Hydroxypyrrol-2(5H)-ones;
The First Total Synthesis of Annomolon A
...36
2.1) General, Regiodefined Access to α-Substituted-Butenolides
...37
2.1.1) Introduction... 37
2.1.2) Current methods for α-butenolide construction... 41
viii
2.1.3.1) Previous work with 3-lithiofurans... 46
2.1.3.2) Optimization work... 52
2.1.3.3)Effect of co-solvent on reactivity of 13... 54
2.1.4) Substrate scope... 58
2.1.5) Ongoing natural product synthesis projects involving 13... 61
2.1.5.1) Isoprotoanemonin as a new building block for α-homoallylic-butenolides... 61
2.1.5.2) Syntheses of labdane diterpenoids... 62
2.2) Lynchpin Strategy Towards Acetogenin Hydroxybutenolides
...66
2.2.1) Introduction to acetogenins... 66
2.2.2) Hydroxybutenolide-containing acetogenins... 72
2.2.3) Model studies for
γ
-hydroxybutenolide acetogenin synthesis... 772.2.3.1) Oxyfunctionalization of silyloxyfurans... 77
2.2.3.2) Model synthesis of anti-inflammatory gorgonian lipid 58... 80
2.3) DMDO-Mediated Oxidative Rearrangement of 2-Furylcarbamates
85
2.3.1) Introduction to γ-hydroxypyrrol-2(5H)-ones... 85
2.3.2) Oxyfunctionalization of 2-silyloxypyrroles... 88
2.3.3) Photooxygenations and autoxidations of 2-furylcarbamates... 91
2.3.4) Development of DMDO-mediated oxidative rearrangement... 93
2.3.5) Deprotection of N-Boc-hydroxypyrrol-2(5H)-ones... 100
2.3.6) Conclusion... 104
2.4) Revised Retrosynthetic Path to Annomolon A
...106
2.4.1) Previous synthetic strategies for trans-mono-THF acetogenins... 106
2.4.2) Novel double-metathesis route to annomolon A... 115
2.4.2.1) Retrosynthesis... 115
2.4.2.2) Stability and reactivity of cyclopropenone acetal 150... 117
2.5) Studies and CM Reactions of γ-Hydroxybutenolide Fragment
...121
2.5.1) Synthesis of butenolide 148... 121
2.5.2) Hydroxybutenolide exploratory cross-metathesis studies... 123
2.5.2.1) Previous butenolide use in CM reactions... 123
2.5.2.2) Model CM reactions with γ-hydroxybutenolide 148... 125
2.6) Synthesis of Mono-THF Fragment for Annomolon A
...129
2.6.1) Setting the stereochemistry by Sharpless asymmetric dihydroxylation... 129
2.6.2) Diastereoselective iodoetherification... 136
ix
2.7) Fragment Unification: Kozmin Double Metathesis Strategy
...143
2.7.1) Initial protecting group-free studies... 143
2.7.2) Resorting to benzyl protection... 150
2.7.3) Chalcogenic misdirection of dienone metathesis... 153
2.7.3.1) Previous examples of chalcogen effects on metathesis events... 153
2.7.3.2) Attempts to block the dienone decoupling of 20... 158
2.8) Completing the Synthesis of Annomolon A (51)
...163
2.8.1) Stepwise enone approach for CM unification... 163
2.8.1.1) Tandem metathesis-hydrogenation strategy... 163
2.8.1.2) Model enone CM coupling of butenolide 148... 167
2.8.2) Completion of annomolon A synthesis... 168
2.8.3) Characterization of synthetic annomolon A... 171
2.8.3.1) Endgame of the synthesis... 171
2.8.3.2) Comparison of natural and synthetic 51... 173
2.8.3.3) Ongoing work: concise synthesis of donnaienin A... 179
2.9) Conclusion
...181
2.10) References
...183
Chapter 3: One Click, Two C=C Bonds:
Enantioselective Total Synthesis of (-)-Auxofuran
...202
3.1) Auxofuran: Origins and Biological Relevance
...203
3.1.1) Streptomyces and its secondary metabolites... 203
3.1.1.1) A rich antibiotic heritage... 203
3.1.1.2) Mycorrhiza... 204
3.1.1.3) Mycorrhiza-helper bacteria... 207
3.1.1.4) Auxofuran... 209
3.1.2) Synthesis of related structures... 212
3.1.2.1) Correction of the original elucidation... 212
3.1.2.2) MMFs... 214
3.1.2.3) (+)-Xestoquinone... 214
3.1.2.4) γ-Hydroxytetralones... 216
3.2) Synthetic Plan: Hetero-Diels Alder/retro-Diels Alder Strategy
...218
3.2.1) Retrosynthesis... 218
3.2.2) Intermolecular HDA-RDA reactions of 4-phenyloxazole... 220
x
3.3) Intramolecular Diels-Alder/Retro-Diels-Alder Approach
...227
3.3.1) Preparation of ynone 44 and diene 20... 227
3.3.1.1) Synthesis of ynone 44... 227
3.3.1.2) Preparation of 4-phenyloxazole (20)... 229
3.3.2) Synthesis of 2-acyloxazoles... 230
3.3.3) Attempts to incite furan formation... 233
3.4) Intermolecular Route:
Synthesis of S-alcohol and “Click” Oxazole Screen
...238
3.4.1) Asymmetric transfer hydrogenation... 238
3.4.2) Confirmation of absolute stereochemistry: synthesis of (R)-γ-Nonalactone... 242
3.4.3) Testing dienophile substrates... 243
3.4.4) Results of diene screen... 245
3.5) Friedel-Crafts Acyl Annulation Work
...248
3.5.1) Precedent for Friedel-Crafts rearrangement... 248
3.5.2) Results for the acid chloride... 252
3.5.3) The mixed anhydride method... 257
3.5.4) Confirmation of Süssmuth’s chiral assignment of auxofuran... 262
3.5.4.1) The advanced Mosher method... 263
3.5.4.2) Auxofuran’s absolute configuration... 265
3.5.5) Conclusion: a simple and enantioselective route to auxofuran... 267
3.6) Click-Oxazole Route to Bioactive Maleic Anhydrides/Imides
...268
3.6.1) Biological activities of maleic anhydrides and imides... 268
3.6.2) Previous syntheses of chaetomellic anhydride A... 271
3.6.3) Rapid click-DA synthesis of chaetomellic anhydride A... 273
3.6.4) A formal synthesis of all Camphorata anhydrides/imides... 275
3.6.4.1) Previous syntheses... 275
3.6.4.2) Original Plan and Initial Results... 277
3.6.4.3) Revised route to relay anhydrides... 280
3.6.5) Alternative synthesis of camphoratas via antrocinnamomin D... 284
3.6.6) Conclusion... 287
xi
EXPERIMENTAL PART
General Information... 303
Chapter 2: Methodologies for the Regiocontrolled Assembly
of α-Butenolides and 5-Hydroxypyrrol-2(5H)-ones;
The First Total Synthesis of Annomolon A
...305
2.1) Syntheses and characterization data for 14, 24, 10a-l, 29, 40... 305
2.2) Syntheses and characterization data for 64, 65, 58, 66... 317
2.3) Syntheses and characterization data for 74c,e,k,l, 75a-k, 78a-j, 80, 82, 83, 84g,j, 85g, 86-94, 95e,g,h,l, 96e,g,h, 97g, 98g 321
2.5) Syntheses and characterization data for 148, 150, 164, 177-184... 350
2.6) Syntheses and characterization data for 146, 149, 151, 185-188, 192, 193, 198, 199, 203-205... 359
2.7) Syntheses and characterization data for 51, 209, E/Z-210, 212-221, 252-255... 371
2.8) Syntheses and characterization data for 152, H/L-266, 267-271, E/Z-273, 274-279, 284... 387
Chapter 3: One Click, Two C=C Bonds:
Enantioselective Total Synthesis of (-)-Auxofuran
...405
3.3) Syntheses and characterization data for 20, 43, 44, 48, 50, 55, 58-64... 405
3.4) Syntheses and characterization data for 70-72, 74-77... 415
3.5) Syntheses and characterization data for 1, 80, 85, 86, 97-99, 103, 104... 425
3.6) Syntheses and characterization data for 105, 111, 112, 128-131, 135-146... 435
References... 453
xiii
LIST OF TABLES
TABLE 1.1: The “E” factor in the chemical industry 22
TABLE 2.1: Initial results involving HMPA as co-solvent 53
TABLE 2.2: Optimization of reaction conditions 54
TABLE 2.3: DMPU effect on reactivity of furan 25 55
TABLE 2.4: Testing co-solvent conditions 56
TABLE 2.5(a): Variety of α-alkylated butenolides (10a-f) synthesized by
optimized procedure 58
TABLE 2.5(b): Variety of α-heteroatom butenolides (10g-l) successfully synthesized 60
TABLE 2.6: Cheng and Xiao’s syntheses of 5-hydroxypyrrol-2(5H)-ones from
2-silyloxypyrroles 88
TABLE 2.7(a): Synthesis of N-Boc-5-hydroxypyrrol-2(5H)-ones 78a-f from 2-furoic
acids 74a-f 94
TABLE 2.7(b): Synthesis of N-Boc-5-hydroxypyrrol-2(5H)-ones 78g-i from 2-furoic
acids 74g-i 95
TABLE 2.8: Optimization attempts for Claisen rearrangement of 187 130
TABLE 2.9: Iodoetherification attempts with IDC-Hex and IDCP 139
TABLE 2.10: Optimization of epoxide-opening of 185 141
TABLE 2.11: Examples from Walkinhshaw group’s study into opening of 206 with
vinylcuprates 142
TABLE 2.12: Catalyst screen for protecting group-free CM reaction 146
TABLE 2.13: Assignment of 1H NMR data for natural annomolon A and 51 175
TABLE 2.14: Comparison of 13C NMR values of THF in 51 to known acetogenins 176
TABLE 2.15: Comparison of bis-MTPA ester data, natural vs synthetic annomolon A 177
TABLE 2.16: Comparison of 13C NMR data for butenolide segment of 51 177
TABLE 2.17: 1H and 13C NMR data of other α-alkylated γ-hydroxybutenolides 178
TABLE 3.1: Conditions used for synthesis of ynone ester 44 228
TABLE 3.2: Conditions used and results of different dienophile reaction with 20 244
TABLE 3.3: Conditions used and results with different dienes 246
TABLE 3.4: Screening of mixed anhydride conditions for FC cyclization 261
TABLE 3.5: Comparison of chemical shifts for (R) and (S)-MTPA of natural and
synthetic 1 266
xv
LIST OF FIGURES
Figure 1.1: Examples of natural product total syntheses by the Woodward group 6
Figure 1.2: A few examples of the many natural product total syntheses by Corey 8
Figure 1.3: Number of FDA-approved drugs in the United States from 1981 to 2007 12
Figure 1.4: The evolution of Eisai’s new breast cancer drug E7389 from halichondrin B 13
Figure 1.5: Jacobsen’s Cr(III)-salen catalysts used in Hetero Diels-Alder reactions 15
Figure 1.6: Number of publications/patents on metal-catalyzed reactions per year 17
Figure 1.7: The quantitative measure for an “ideal” total synthesis 18
Figure 1.8: Difference between atom economy and reaction yield 23
Figure 2.1: α-substituted butenolides 37
Figure 2.2: Existing 3-carbanion approaches to α-butenolide construction 44
Figure 2.3: Methods for invoking lithium-halogen exchange of 3-halosilyloxyfurans 48
Figure 2.4: Disaggregation effect of polar solvents leading to enhanced carbanion
nucleophilicity 51
Figure 2.5: The leoheteronins and the labdane structural unit 62
Figure 2.6: The acetogenin blueprint and seven classes based on THF/THP pattern 67
Figure 2.7: Postulated biosynthetic routes to acetogenins from polyketides 68
Figure 2.8: Purported biological mode of action of acetogenins 69
Figure 2.9: “Ca2+ shuttling” role of acetogenins, increasing mitochondrial *Ca2+] 70
Figure 2.10: Tautomerism and possible biological interactions of
γ-hydroxybutenolide acetogenins 76
Figure 2.11: Cytotoxicity data (IC50) of myceliothermophins A-D against cancer cells 90
Figure 2.12: Super-Grignard generation using lithium chloride 97
Figure 2.13: Commercially-available ruthenium-based catalysts for metathesis 113
Figure 2.14: Cyclopropenones produced by nature 118
Figure 2.15: Intermolecular cycloadditions reactions of singlet π-delocalized
vinylcarbene 153 118
Figure 2.16: Catalytic cycle of Sharpless AD 132
Figure 2.17: (a) Corey’s postulated U-shaped binding mode (b) Sharpless mnemonic 133
Figure 2.18: Popular iodonium (I) sources 138
Figure 2.19: Hoye’s first demonstration of allylic hydroxyl activation of RCM 153
Figure 2.20: Recent examples of other acetogenin syntheses yielding
different optical rotations 173
Figure 3.1: Well-known antibiotics originating from Streptomyces 204
Figure 3.2: Interactions in the rhizosphere among plants, mycorrhizal fungi, and
bacteria 206
Figure 3.3: Mechanisms for rhizosphere bacteria to adhere to the cell surface of
mycorrhizal fungi, causing a change in fungal gene expression 210
xvi
Figure 3.5: FC acylations of t-butyl esters catalyzed by indium-tribromide 249
Figure 3.6: Wissner’s HCl-free conversion of silyl-carboxylates to acid chlorides 251
Figure 3.7: Illustration of anisotropic effects in dominant MTPA ester conformers 264
xvii
LIST OF SCHEMES
Scheme 1.1: Robinson’s 1917 total synthesis of tropinone 5
Scheme 1.2: Belanger’s Vilsmeier-Haack-azomethine ylide cascade for synthesis
of Daphnane tricycle 10
Scheme 1.3: Two possible pathways for HDA reaction of carbonyl compounds 16
Scheme 1.4: Biosynthesis of penicillins 19
Scheme 1.5: The Leighton synthesis of zincophorin and its ester 20
Scheme 1.6: Rapid polyketide assembly by tandem silylformylation crotyl-silylation 21
Scheme 1.7: An epoxide-opening cascade promoted by water 24
Scheme 1.8: The azide-acetylene “click” reaction 25
Scheme 1.9: Fluorescent in-vivo biomolecular labelling made possible by
copper-free click chemistry 26
Scheme 2.1: Rawal’s and Trauner’s synthetic strategy to the bipinnatins and related
targets 40
Scheme 2.2: Recent methods for α-butenolide synthesis from acyclic precursors 42
Scheme 2.3: Recent methods for α-butenolide synthesis from oxacyclic precursors 43
Scheme 2.4: Other recent methods for α-butenolide synthesis from oxacyclic
precursors 44
Scheme 2.5: Kuwahara’s α-substituted butenolide synthesized via sulphoxide
elimination 45
Scheme 2.6: Jones’ method to α-butenolides via flash vacuum pyrrolysis 45
Scheme 2.7: Our proposed strategy compared to the Näsman protocol 46
Scheme 2.8: Yields for electrophilic quench of 12 compared to 2-PON-furan 47
Scheme 2.9: The CIP/SIP pathways for an SN2 reaction of an organolithium 57
Scheme 2.10: Possible effects of solvation on lithium counterion role 57
Scheme 2.11: Faulkner’s photoxygenation approach to γ-hydroxybutenolides 77
Scheme 2.12: Mechanistic pathways for DMDO-mediated route to
γ-hydroxybutenolides 79
Scheme 2.13: Original retrosynthesis for annomolon A 81
Scheme 2.14: Kim’s synthesis of 5-hydroxypyrrol-2(5H)-ones from
γ-alkylidene-butenolides 87
Scheme 2.15: Mechanistic scenarios for DMDO reaction of 2-silyloxypyrroles 68 89
Scheme 2.16: Photooxygenations (method A) and autoxidations (method B) of
2-furyl-carbamates 91
Scheme 2.17: Photooxygenation studies of activated and deactivated
2-(5-aryl)-furyl-carbamates 92
Scheme 2.18: Proposed mechanism for our DMDO-mediated oxidative
xviii
Scheme 2.19: The DMDO-directed synthesis of azepine-2,5-dione 80 via 78f 98
Scheme 2.20: Revised retrosynthesis for the total synthesis of annomolon A 116
Scheme 2.21: Planned route to epoxide 185 129
Scheme 2.22: Chair transition states for Claisen rearrangement of vinyl ether of
187 130
Scheme 2.23: Depiction to explain preference of trans-THF formation between 199
and IDCP 137
Scheme 2.24: Lemieux’s procedure for preparing iodonium dicollidine perchlorate 138
Scheme 2.25: Tius’ Nazarov cyclizations catalyzed by silica gel 145
Scheme 2.26: Possible pathway for decoupling of dienone from 209 and formation
of 212 148
Scheme 2.27: The influence of different OR allyl groups on ring size selectivity 155
Scheme 2.28: Homoallylic directing effect leading to CM-favoured 228 en route
to (+)-neopeltolide 156
Scheme 2.29: Illustrative examples from Kozmin’s dienone CM reactions 159
Scheme 2.30: The ideal, protecting group-free plan for utilizing 152 for fragment
unification 164
Scheme 2.31: Grubbs’ three-step tandem sequence for the synthesis of
(R)-(-)-muscone 165
Scheme 2.32: Formation of active hydrogenation complexes from G1 and G2 165
Scheme 2.33: Retrosynthesis for facile route to donnaienin A 180
Scheme 3.1: Hetero-Diels-Alder/retro-Diels Alder reaction of 4-phenyloxazole
and acetylenes 221
Scheme 3.2: Triethylamine-catalysed oxetane formation and ketene dimerization 229
Scheme 3.3: Proposed mechanism of Noyori transfer hydrogenation 240
Scheme 3.4: Literature precedent for various FC tetralone-forming scenarios 248
Scheme 3.5: Possible activation routes for desired FC rearrangement 250
Scheme 3.6: Acid-catalyzed racemization of (S)-lactone in synthesis of trecetilide
hemi-fumarate 250
Scheme 3.7: Mechanistic pathways for lactonization of 80 or 86 256
xix
ABBREVIATIONS
acac: acetoacetonate AIBN: azobisisobutyronitrile AMP: adenosine monophosphate ATP: adenosine-5'-triphosphate BARAC: biarylazacyclooctynone BHT: 2,6-di-tert-butyl-4-methylphenol
BINAP: 2,2'-bis(diphenylphosphino)-1,1'-binaphthyl BJAB: Burkitt’s lymphoma cell line
Boc: tert-butyl carbamate Bn: benzyl
CBS (reagent): Corey-Bakshi-Shibata oxazaborolidine reagent CoA: coenzyme A
COD: cyclooctadiene
COX-1/2: cyclooxygenase 1/2 Cp: cyclopentadiene
CSA: camphorsulfonic acid Cy/cy: cyclohexyl dba: dibenzylideneacetone DBP: dibutyl phthalate DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene DCC: N,N'-dicyclohexylcarbodiimide DCE: 1,2-dichloroethene DDQ: 2,3-dichloro-5,6-dicyano-1,4-benzoquinone DIBAL-H: diisobutylaluminium hydride
DIFO: difluorinated cyclooctyne DIP: diisopinocampheyl borane
DIPEA: diisopropylethylamine (Hünig’s base) DMAD: dimethyl acetylenedicarboxylate DMAP: 4-(N,N-dimethylamino)pyridine DMDO: dimethyldioxirane dmgH: dimethylglyoxime DMF: N,N-dimethylformamide DMP: 2,2-dimethoxypropane DMPU: 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone DMSO: dimethylsulfoxide DMT: N,N-dimethyltryptamine DNA: deoxyribonucleic acid
DTBMP: 2,6-di-tert-butyl-4-methylpyridine E: electrophile
ED50: half maximal (50%) effective dose
xx
GI50: half maximal (50%) growth inhibitory concentration
HMPA: hexamethylphosphoramide
HMBC: heteronuclear multiple bond correlation (spectroscopy) HMDS: hexamethyldisilazane
HPLC: high performance liquid chromatography IC50: half maximal (50%) inhibitory concentration
IDC-Hex : iodonium bis(sym-collidine) hexafluorophosphate IDCP : iodonium bis(sym-collidine) perchlorate
IMDA: intramolecular Diels-Alder Imid.: imidazole
KHMDS: potassium hexamethyldisilazide LAB: lithium diisopropylaminoborohydride LDA: lithium diisopropylamide
mCPBA: meta-chloroperoxybenzoic acid
Mes: mesityl (1,3,5-trimethylbenzene) MMPP: magnesium monoperoxyphthalate
modp: (Z)-2-hydroxy-5,5-dimethyl-1-morpholinohex-2-ene-1,4-dione MOM: methoxymethyl ether
Ms: methylsulfonyl
MTPA: α-methoxy-α-trifluoromethylphenylacetate NA: not determined/available
NADH: nicotinamide adenine dinucleotide (reduced form) NaHMDS: sodium hexamethyldisilazide
NBS: N-bromosuccinimide NMP: N-Methyl-2-pyrrolidone NO: nitric oxide
PCC: pyridinium chlorochromate PDC: pyridinium dichromate PG: protecting group
Piv: pivalyl (trimethylacetyl) PMB : para-methoxybenzyl PPA: polyphosphoric acid PPi : pyrophosphate
ppm: parts per million
PPTS: pyridinium p-toluenesulfonate py: pyridine
ROMP: ring-opening metathesis polymerization rt: room temperature
sym-collidine: 2,4,5-trimethylpyridine
TBAF: tetrabutylammonium fluoride TBHP: tert-butyl hydroperoxide TBS/TBDMS: tert-butyldimethylsilyl
TEMPO: (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl Teoc: trimethylsilylethyl carbamate
xxi
Tf: trifluoromethylsulfonyl TFA: trifluoroacetic acid
TFAA: trifluoroacetic anhydride THF: tetrahydrofuran
THP: tetrahydropyran TIPS: triisopropylsilyl
TLC: thin layer chromatography TMS: trimethylsilyl
TPAP: tetrapropylammonium perruthenate TPS/TBDPS: tert-butyldiphenylsilyl
Ts: para-toluenesulfonyl
CHAPTER 1:
3
1.1) Introduction to the Total Synthesis of Natural Products
1.1.1) The beginnings of natural product synthesis
Throughout the twentieth century, the broad ranging study of atomic properties, behaviour and reactivity that has been historically qualified as the science of chemistry has played an important role in shaping the technological advances of our world. These advances have underpinned all the changes, mainly positive but sometimes inadvertently negative, that our societies have undergone in that time. In this sense, organic synthesis has been one of the most expressive exponents of this science, owing to its creative power and seemingly limitless scope. To gauge the impact of synthetic organic chemistry on the modern world it suffices only to look around us and acknowledge its key role in pharmaceuticals, fertilizers, pesticides, fuels, high-tech materials, polymers, cosmetics, clothing, and more recently nanohigh-technology.1 Arguably a vital driving force behind the progress achieved in organic synthesis has been the practice of total synthesis of natural products. Few other scientific endeavours have embodied more the notion that “necessity is the mother of invention”. This discipline, through its primary goal of constructing the most structurally complex and challenging of nature’s molecules, has over the decades served to channel and focus efforts at inventing new chemical reactions.2 In this way the field of total synthesis has led to a greater understanding and prediction of chemical reactivity and selectivity, and continues to take pride of place as the ultimate testing ground for new methodologies and strategies.1
Total synthesis is precisely defined as the complete, chemical elaboration of complex organic
molecules from simpler precursors, solely by a series of manmade chemical reactions.3 The simpler starting materials are ones that are inexpensive and available in large quantities, often being the products of petrochemical processes, but can also have natural origins such as sugars and amino acids. The first true total synthesis in the sense of these words was therefore Friedrich Wöhler's 1828 synthesis of urea, which despite the trivial nature of the molecule was seen as a landmark for various reasons.4 Firstly it demonstrated that organic molecules can be produced from inorganic precursors, in this case ammonium cyanate, and secondly it helped disprove the still persistent theory of vitalism.5 This notion that there was an innate force
4
distinguishing biological processes from non-living chemical ones, in short the belief in life emanating from a mysterious vital force, or “soul”, was even held by such eminent scientists as Louis Pasteur. In its early days, total synthesis thus held a profoundly significant role in demystifying life-giving biochemical reactions by showing that man could replicate the molecules nature produced. The synthesis of acetic acid from elemental carbon by Kolbe in 1845 was also historically significant as it marked the first use of the word “synthesis” to describe the process of assembling a chemical compound from other substances.6 The powerful consequences on society of pursuing natural product synthesis are epitomized by Perkin’s serendipitous synthesis of mauveine in 1856, at the tender age of 18.7 Whilst taking on the synthesis of the antimalarial quinine in a challenge set by his professor, Perkin attempted the triple oxidation of N-allyl toluidine. The unexpected product, known as aniline purple, led to large scale dye manufacturing as a lucrative business, and is widely considered to represent the birth of the chemical industry.7b
The list of natural products achieved by total synthesis continued to grow, with most targets chosen for their similarity to available starting materials and thereby being attainable through a series of simple functional group conversions. A true landmark came in 1890 with Fischer’s synthesis of (+)-glucose, which represented a quantum leap forward at the time in terms of the complexity of the target structure. With its five proximal chiral centres arranged in a single hexacycle this was truly a challenging natural product for a chemist of that day to have constructed, earning Fischer the Nobel Prize for chemistry in 1902.8 Other notable total syntheses of the pre-war era include those of camphor by Perkin (1904) and Komppa (1903)1, who successfully industrialized his process, α-terpineol by Perkin (1904)9, and tropinone by Willstätter (1901)1 and Robinson (1917).10 Robinson’s route, illustrated in Scheme 1, was particularly impressive as it brought together three components, succindialdehyde,
5
methylamine, and acetone dicarboxylic acid by a cascade of intramolecular and intermolecular Mannich reactions, all in a single step.10
Scheme 1: Robinson’s 1917 total synthesis of tropinone10
1.1.2) Maturing of a discipline: the Woodwardian era
Up until the early 1930s the role of natural product synthesis was very much an analytical one of structural determination and confirmation, since there were few better tools at the time for elucidating a molecule’s identity. As organic chemistry grew into a field in its own right, the natural products made accessible to chemists became gradually ever more complex. This increased complexity was inherent with scarcity, therefore giving total synthesis the added valuable role of providing easier and preferably cheaper access to biologically and medicinally important compounds. This helped evolve the standing of total synthesis from being merely a molecular proofreading exercise to more of a sophisticated art form.11 In short, natural product synthesis went from being a showcase for new organic reactions being discovered, to an actual tool through which such discoveries could be made. This was important as it catapulted total
6
synthesis from the periphery of chemical research to the centre stage where it would attract some of history’s most brilliant scientific minds.
Figure 1: Examples of natural product total syntheses by the Woodward group12
One such individual, R. B. Woodward, provided much of this impetus, transforming organic synthesis both into a highly rational science adhering to strict principles of physical chemistry, and a sophisticated art demanding elegant solutions. He did more than any other to influence and contribute to the domain of total synthesis in the four decades of his career, and revolutionized the field of structural determination. Through meticulous planning, and at an incredible speed given the infancy of spectroscopic methods compared to today, many impossibly complex natural products were synthesized in his group, including quinine, cortisone, strychnine, lysergic acid, reserpine, chlorophyll α, and cephalosporin C (Figure 1).12 An important aspect of his work was the use of rings to install and control stereochemical centres, from which further functionality could be unravelled through their rupture, a tactic that have become standard fair today.1 Organic chemistry was fundamentally changed by his work, most significantly in the development of the Woodward-Hoffmann rules on conservation
7
of orbital symmetry.13 His total synthesis of (-)-strychnine in 1954 exemplified the state of the art with its conciseness, attention to detail, and powerful stereoselective conversions.12c Along with Eschenmoser, in 1973, his group published arguably the most colossal natural product synthesis of all time, that of vitamin B12. It took over a decade to complete, and it is noteworthy
that since then no other synthesis has been published.14
The role of torchbearer for Woodward’s mantle was informally handed down to E. J. Corey, who ushered in an era of greater logic through his spreading of the practice of retrosynthetic analysis. This systematic approach to complex structures consisted of breaking them down into ever more simple organic precursors, or synthons, by a series of strategic bond disconnections.15 This formalization of a way of thinking that was already practised in the minds of most synthetic chemists, made the task of total synthesis more efficient and engendered practitioners with routes that were more flexible in terms of the synthetic equivalents that could be employed. Corey not only pioneered this approach, proving its worth via an enormous portfolio of successfully attained natural product targets (Figure 2)16, but also the concept of Umpolung (along with Seebach)17, and is credited with the discovery of many useful synthetic reactions and methodologies. He was particularly active in the area of asymmetric synthesis, and the development of boron-based asymmetric catalysis, such as the Corey-Bakshi-Shibata oxazaborolidine reagent.18
As well as Corey and Woodward the field of total synthesis has benefited from the groundbreaking contributions of many innovative and resourceful chemists, such as Brown, Stork, Eschenmoser, Djerassi, Bartlett, Nicolaou, Danishefsky and Ley to name but a few.1,2,11 Standing on the shoulders of these giants, contemporary synthetic organic chemists are now
8
blessed with a mindboggling array of chemical reactions that means no known natural product, irrespective of its structure, is theoretically out of reach.
9
1.2) The Utility of Total Synthesis
1.2.1) Structural determination and new chemistry development
As mentioned in the previous pages, the role of total synthesis evolved from its early inception as a way of linking life-permitting biochemical processes to the fundamental physical laws that govern atoms in all living and non-living objects, into the ultimate turn of the century tool for accurate structural elucidation. Since then, the advent of more powerful, sophisticated analytical techniques, particularly NMR spectroscopy and X-ray crystallography, has made total synthesis take more of a backseat in structural determination. This however does not render it obsolete for this purpose, and there are still almost daily reports published in which proposed structures are revised either subtly or significantly by a successful total synthesis. It goes without saying that it is still the only way to unequivocally confirm structure and stereochemistry, and that a natural product’s structure tends to remain tentative in the literature until a group of chemists somewhere in the world has given the synthetic nod of approval.
As a noble endeavour in its own right, the intellectually stimulating task presented to a budding student of organic chemistry by the challenge of synthesizing a compound with a totally new structure merits academic pursuit. Investment, both mental and material, by society into this area of work is amply rewarded by the chemical and analytical inculcation it bestows on the practitioner. In the same way that teaching a subject is the most effective way for somebody to learn it, the same can be said of organic reactions. No chemist would argue that an organic reaction was better understood from listening to a lecturer or reading a textbook than it was from having to depend on it for a synthesis project.
Without negating this pedagogical perspective however, the greatest importance of total synthesis comes in its use as a platform for developing innovative strategies and discovering new chemistry. In so doing, this enables not only a target structure to be accomplished but can also offer novel solutions to more profound chemical problems of general interest to the scientific community. Examples abound of total synthesis projects opening such doors onto
10
new organic reactivity, such as Sheehan’s work in the 1950s in elaborating the β-lactam ring of penicillin, which led to the invention of carbodiimide-based reagents that revolutionized peptide bond formation.19 The wealth of knowledge that can be found in textbooks on the synthesis of heteroaromatic systems and the unique physical properties of quinoline and piperidine rings, owes much to the continuous efforts directed toward the synthesis of quinine.1,11 In the 1960s and 1970s, the projects undertaken in Corey’s group towards the prostaglandins inspired the creation of the first catalysts for asymmetric Diels–Alder reactions11,16a, as well as the development of the now familiar silyl-based protecting groups that no chemist nowadays can do without.20 More recent reactions owing their existence to total synthesis pursuits include Evans’ pioneering use of oxazolidinone chiral auxiliaries for asymmetric aldol reactions21, Movassaghi’s cobalt-catalyzed cyclotryptamine dimerizations22, and the Bélanger group’s intramolecular Vilsmeier-Haack-azomethine ylide cycloaddition cascade for the synthesis of the tricyclic daphnanes (Scheme 2).23
Scheme 2: Bélanger’s Vilsmeier-Haack-azomethine ylide cascade for synthesis of Daphnane tricycle23
1.2.2) Biological importance
Natural product synthesis is also a vital element in the advancements that have been made in biology and medicine.1,19,24 In the early 1900s, 80% of all medicines were obtained from roots, barks and leaves, and more recently, natural products have continued to be significant sources of drugs and leads. Their dominant role is evident in the approximately 60% of anticancer compounds and 75% of drugs for infectious diseases that are either natural products or natural product derivatives.25 When natural products of significant medical import, such as taxol or erythromycin, cannot be obtained easily through isolation, total synthesis is often the only
11
viable option to obtaining the product in the multigram quantities normally required for a thorough study of its biological profile. Academic syntheses can then also offer an industrially scalable chemical pathway to these compounds if they become medicines, or provide a malleable route for process chemists to optimize. Furthermore, an expedient total synthesis of a natural product can eventually be modified to provide analogues possessing superior biological activity or diminished nefarious effects.
Natural product synthesis and drug development go hand in hand. Luckily for us, nature holds a billion year head start over our own chemical knowledge in terms of designing molecules that can impart beneficial biological effects against disease states. These molecules are normally involved in complex and subtle interactions with receptive sites on proteins in the cells of organisms, causing a modification in biochemical cascade sequences that eventually transpires in the desired medicinal outcome. Very similar enzymatic processes have worked in cohort to produce these unique compounds in the plants and animals they have been isolated from, not to mention the role their molecular frameworks play within those organisms in interacting and binding to specific receptor sites.26 This logically means that natural products are much more likely than any other organic structure to exhibit some sort of biological activity when taken up by the cells of another organism, such as a bacteria, mouse or human. Historically, the statistics back up this rationale: if one considers only polyketide metabolites, just over 7000 known structures have led to more than 20 commercial drugs, with a “hit rate” of 0.3%, which is much better than the <0.001% hit rate for high-throughput screening (HTS) of synthetic compound libraries.27
This fact has been somewhat lost over the last two decades of pharmaceutical research (see Figure 3), and although more than 100 natural product–based drugs were in clinical studies as of 2009, this represents a nearly 30% drop from the years 2001 to 2008 inclusive.27 Many pharmaceutical firms in fact have terminated entirely their natural product programs, turning increasingly to combinatorial chemistry, bioinformatics and computational design28 as a way to try and eliminate the trial and error process associated with natural product-derived drug
12
design. This new zeitgeist in big pharma however has not led to the expected exponential leap in lead compound discovery despite the efficacy of HTS techniques, in particular in the key therapeutic areas of immunosuppression, anti-infectives, and metabolic diseases. Moreover, it is becoming increasingly clear that the most challenging malaises still to be effectively tackled by the industry will require the invention of drugs that can interact with multiple receptors and/or enzymes, and not just the single binding sites that mass screening of small, often flat, aromatic molecules is geared towards.29
Figure 3: Number of FDA-approved drugs in the United States from 1981 to 2007.27
In this regard many natural product structures, such as those of large polyketide macrocycles or polycyclic three-dimensional alkaloids, can offer renewed platforms for multiple phenotype screening. The very recent successful development of anticancer analogues of halichondrin B serves as a superb case against the prevailing dogma in pharma that the total synthesis of complex natural products is purely an academic exercise. Following Kishi’s elegant total synthesis of halichondrin B in 199230, the Eisai Research Institute pursued further medicinal studies of this highly complex marine metabolite and honed in on the pharmacophore responsible for its tubulin-inhibiting activity. As Figure 4 shows, the medicinally-relevant segment of halichondrin B could be replicated by simpler analogues, which were synthesized by
13
sticking to Kishi’s original convergent strategy.31 The Eisai team was able to prepare a series of highly potent leads, resulting in the selection of E7389 for further development (Figure 4). As of November 2010, E7389 (now eribulin mesylate or Halaven) has been approved by the US Food and Drug Administration for the treatment of metastatic breast cancer.31b
Figure 4: The evolution of Eisai’s new breast cancer drug E7389 from halichondrin B30,31
1.3) Modern Total Synthesis
1.3.1) The holy trinity: chemo-, regio- and stereoselectivity
The state of the art in contemporary organic synthesis has reached such a refined level of sophistication that there is no longer a molecule, as Baran put it, “that appears hopelessly complex”.32 Indeed, the sense of hope that was part and parcel of many total synthesis projects twenty years ago, has steadily been replaced by one of impatience, as more and more finely tuned organic methodologies have been developed to solve whatever structural or functional conundrum a target compound may pose. The holy grail of nearly all modern organic synthesis projects is that of selectivity. In order to successfully build more complex, highly functionalized structures, a great deal of the same reactions of the past have been revisited and in many cases even resurrected, but with new chemo and regioselective reagents expressly developed to perform these transformations with minimal perturbation elsewhere. Oxidation reactions are a perfect example, with a myriad of procedures and reagents that now exist such as Corey’s PCC reagent, Dess-Martin periodinane, Ley’s TPAP and many more.33 An important theme in this thesis is the utilization of dimethyldioxirane (DMDO) in chapter 2 for milder, more
14
regioselective oxyfunctionalizations of furans and pyrroles, which has permitted syntheses that would otherwise have been impossible with stronger oxidizers such as mCPBA or peroxides.34
By far the most invested area in the search for selectivity has been in the harnessing of highly asymmetric, catalytic organic reactions, with the emphasis on carbon-carbon bond forming processes.1,11,19,35 Aldol reactions, with an eye on polyketide synthesis, have been exhaustively subjected to enantioselective upgrading.36 In general, control of the relative and absolute configuration of the newly formed stereogenic centers has been achieved through the use of chiral starting materials or chiral auxiliaries. The activation of the acceptor carbonyl with chiral ligands bound to Lewis acids such as tin, titanium, iron, copper and gold has been very successful for reactions with silyl enol ethers, known as the Mukaiyama aldol.37 The closely related asymmetric allylations of aldehydes have seen some ingenious methods invented, most notably Brown’s pinene reagents and the Sakurai reaction.38 Acetylenes have not been forgotten either, with Carreira’s asymmetric aldehyde alkynylation having become a staple of many synthetic projects.39 An immensely influential asymmetric reaction of the past twenty years has been the Sharpless asymmetric dihydroxylation (AD), which is a key feature of the chemistry elaborated in this thesis (chapter 2). This chiral modification of the Upjohn reaction, allows cis-hydroxylation of olefins with superb levels of enantioselectivity. The reaction is very useful as it allows the imparting of chirality onto unsaturated hydrocarbon backbones in a similar fashion to how enzymes in nature oxidize double bonds, thereby setting the stage for further stereogenic creation as well as providing a functional handle that can be easily exploited for subsequent chemistry.40 This is amply demonstrated in the proceeding chapter, where the Sharpless AD is used as a lauchpad for creating the four chiral centres of the antitumor acetogenin, annomolon A.41
Diels-Alder chemistry is also one of the most substantially advanced and researched areas of modern asymmetric synthesis, owing to its capacity for multiple carbon-carbon bond creation in a single simple operation.42 The steady advancement of catalytic variants that lessen the need for the traditionally high temperatures associated with this pericyclic process has made
15
them more amenable to sensitive, intricate structures. The tactic of employing Lewis acids to activate dienophiles enhances the normal demand Diels-Alder reaction since the gap between the lowest unoccupied molecular orbital (LUMO) of the dienophile and the highest occupied molecular orbital (HOMO) of the diene is reduced. Another substantially elaborated strategy, this time centred on the diene, has been the use of Danishefsky’s diene to engender improved regioselectivity.43 The success here emanates from the position of the two oxygens on the diene that cooperate to electronically direct regiospecific formation of a lone endo adduct when reacted with most dienophiles.
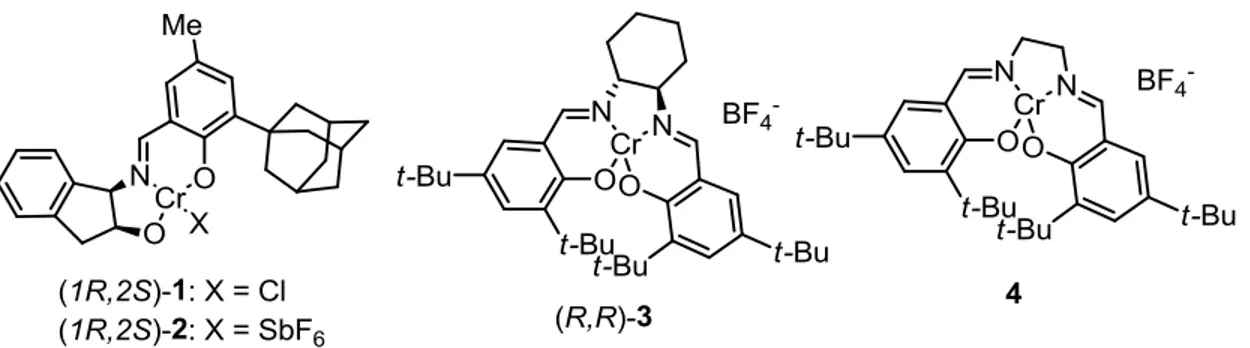
Figure 5: Jacobsen’s chiral Cr(III)-salen catalysts used in Hetero Diels-Alder reactions44
This has been put to good effect in hetero Diels-Alder reactions with aldehydes, and has allowed the development of a powerful asymmetric protocol that employs chiral chromium catalysts (see Figure 5).44 This work from the Jacobsen group has already seen some elegant applications, and was initially considered for the total synthesis detailed here in chapter 2. These tridentate complexes (1-4), such as 1 that was used in the key reaction for the synthesis of fostriecin44d, permit the direct formation of chiral compounds from achiral substrates under mild conditions. As Scheme 3 indicates, there are two possible pathways that have been identified for the reaction to proceed by, either via a Mukaiyama aldol-formed intermediate, or as is expected from our envisaged conversion of 5 to 6, through a direct [4+2] pericyclic mechanism.44a Notwithstanding these advances, a more generally-applicable catalytic system for asymmetric Diels-Alder reactions is still elusive, and there have been other commendable strategies developed that exploit homochirality on the substrates themselves as a means to engineering the absolute stereochemistry of adducts.42
16
Scheme 3: Two possible pathways for HDA reaction of carbonyl compounds44
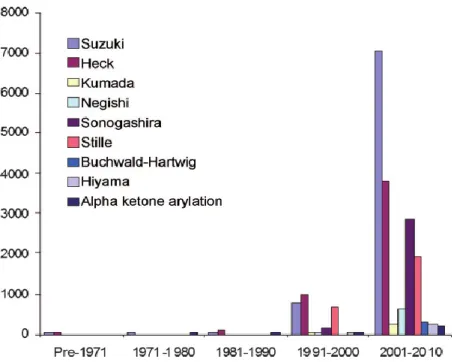
1.3.2) Cross-coupling
One element that has become synonymous with contemporary organic chemistry is palladium, and in fifty years or so future generations of chemists will most likely distinguish this epoch from the previous century of synthetic discovery by referring to it as the era of palladium cross-coupling. Since the Nobel award-winning pioneering work of Heck, Suzuki and Negishi, palladium compounds have been further developed and refined to dizzying heights, and can now catalyze a remarkably versatile range of cross-coupling reactions such as carbon– heteroatom coupling, α-arylation, direct arylation by C-H activation, and decarboxylative coupling. As is evident from Figure 6, a veritable boom in this area has occurred in the last decade in terms of publications and patents, with the Suzuki–Miyaura cross-coupling proving by far the most popular, followed by the Heck and Sonogashira coupling reactions.45 The weak carbon-bonding ability of palladium was apparent early on in its heterogeneous application on charcoal supports for olefin saturations, and the Wacker oxidation that emerged in the 1950s38, but it was not until the early 1970s that researchers began exploiting this for homogeneous reactions. The discovery that palladium (0) would insert oxidatively across carbon-halogen bonds and reductively eliminate after unifying this to another metalated species, has made mild carbon-carbon coupling possible. There are many reviews that detail the historical developments and importance of this chemistry, suffice to say that it has transformed the organic domain, allowing the now trivial incorporation of previously challenging sp2-sp, sp2-sp2, sp2-sp3 carbon coupling manoeuvres into total synthesis projects. The very latest developments
17
in this area aim to couple ever more challenging substrates under milder conditions, with lower palladium catalyst loadings, and using more efficient catalytic systems that make use of a seemingly endless variety of ligands to impart beneficial steric and/or electronic effects.45 The study of ligands especially has been highly influential in opening up the possibilities for newer cross-coupling reactions, most notably the C-N forming Buchwald-Hartwig protocols. The next great challenge that is already being met head-on by researchers such as Du Bois, Hartwig and Yu is that of C-H activation through palladium catalysis.46 The advantage of such an approach is plain to see, with an increased atom economy resulting from the prospect of bypassing derivitization or pre-activation of coupling partners (i.e. as halides or boronates).
Figure 6: Number of publications/patents on metal-catalyzed reactions per year45
The other cornerstone of carbon-carbon cross-coupling chemistry that has only emerged recently but is now seemingly indispensible to modern day synthesis, is that of metathesis. This was a reaction that was rediscovered in a very real sense by academia, since ill-defined tungsten-based metathesis systems were already established for ethylene polymerizations in the petroleum industry.48 After the initial mechanistic revelations of Chauvin and Katz in the
18
1970s47, Schrock developed highly reactive tungsten and molybdenum-based alkylidene catalysts to carry out efficient metathesis reactions.48 These catalysts, though highly reactive and unstable, marked a turning point in the science that signalled the beginning of metathesis applications in total synthesis. Grubbs’ contribution was to develop practical ruthenoid catalysts that were air stable, easy to handle, yet maintained a good enough level of reactivity to be widely applicable in ring-opening metathesis polymerizations, ring closing and cross-coupling metathesis reactions.49 The first and second generation Grubbs catalysts are now the most utilized in modern day applications, and the literature is becoming increasingly populated by other ruthenium catalysts based on this template. As Fürstner summarized in his recent review, the “power of metathesis” for total synthesis can be gauged by the high levels of regio- and chemoselectivity that have been achieved and the exceptional functional group tolerance of the Grubbs’ catalysts.50 As well as this the reactions are reversible and thus flexible, atom economical with no inorganic side-products, and make use of double bonds that are one of the easiest and cheapest functionalities to install.
1.3.3) Working towards the ideal, greener synthesis
1.3.3.1) The concept of “ideality”
Figure 7: The quantitative measure for an “ideal” total synthesis32
With the outstanding synthetic techniques on offer these days for the pursuit of total synthesis of natural products, the onus has shifted away from being able to merely reach the target in some elegant, original fashion, to one demanding heightened economy, both in terms of number of steps, the degree of side-products (atom economy) and oxidation state changes. In the twenty-first century total synthesis is faced with the crucial challenge of being able to provide quickly, at low cost and with the least environmental impact, large quantities of complex natural products. This is especially poignant given the aforementioned drawbacks that
19
are obliging the pharmaceutical sector to turn back to naturally-derived structures for drug inspiration.28,29
Scheme 4: Biosynthesis of penicillins32
A concept that has come to the fore in very recent times is that of the “ideal” synthesis, which draws its inspiration from and uses as a reference point the ability of nature to perform the biosynthesis of natural products in the most efficient manner imaginable. Baran and co-workers were able to give a tangible, quantitative expression to measure the ideality of a synthetic route (Figure 7) by dividing the number of useful, constructive steps that actually contribute to the elaboration of the target structure, by the number of total reactions in the sequence.32 By this yardstick, many total syntheses previously considered proficient, such as Rainier’s very recent brevenalwork (38 steps, 0.99% overall yield), tend to fall by the wayside.51 Indeed the majority of natural products accomplished over the last two to three decades have depended heavily on protection and deprotection steps as well as the use of masked equivalents of the desired functional groups that then necessitate wasteful interconversion steps.2,3,11 As a wonderful comparison, Baran points to the 100% ideality typical of biosynthetic routes to natural products by an illustration of how nature makes penicillin (Scheme 4). Starting with the
20
amino acids cysteine, valine, and amino-adipate, a construction step leads to tripeptide 7, and then a strategic redox reaction follows in which isopenicillin-N-synthase fuses the bicyclic penam scaffold of isopenicillin N. Another construction step provides the eventual structure 8 that is common to all penicillins.32
Scheme 5: The Leighton synthesis of zincophorin and its ester52
Efforts are already being made by synthetic chemists to meet these modern demands; as a case in point, the Leighton lab recently published a total synthesis of zincophorin (Scheme 5) and its methyl ester that surpassed the previous routes by Danishefsky, Cossy and Miyashita in terms of step economy, overall yield and ideality.52 The core of their strategy was the group’s tandem silylformylation allyl(crotyl)-silylation reaction (Scheme 6), a powerful tool for rapid polyol construction.53 This is emblematic of a growing trend in the community that aims to tackle the next great obstacle for synthesis of generating a high degree of stereogenic complexity in the minimum number of steps. In this example the compact, trioxygenated stereo-pentad 9 is constructed with almost ruthless expediency, by exposing silylated alcohol 10 to a very low catalytic amount of bis-carbonylated rhodium under a carbon monoxide atmosphere.52 This effected concomitant silyl and CO insertion across the triple bond of 10, followed up by in situ highly enantioselective crotylation of the intermediate aldehyde 11 to form 12. The protic conditions of the ensuing Tamao oxidation also led to a stereoselective enol tautomerization
21
that gave the product diol 9 in an excellent yield for such a complicated cascade of events.52 The in situ crotylation of 11 hinges on the concept of “strain-release Lewis acidity”53, since silicon itself is normally not acidic enough to activate the carbonyl function. In 11 the silicon is constrained in a tetrahedral silacycle that by coordinating to the proximal aldehyde and thus switching to a trigonal bypyramidal pentacycle leads to a lowering in energy due to strain release.54 This enhancement of the silicon’s acid character was coined strain-release Lewis-acidity by Denmark55, and has also been exploited by Leighton for asymmetric allylations/crotylations using newly developed chiral allyl/crotyl-diazasilane reagents.56 These reagents offer some advantages over the well-established Brown boron-based methods38 that suffer from drawbacks such as instability and laborious preparation or workup procedures. These silanes are crystalline and air-stable, can be prepared in high yield and purity and on large scales.56
Scheme 6: Rapid polyketide assembly by a tandemsilylformylation crotyl-silylation reaction55,56
This exploitation of a new facet of silicon chemistry by one group for more efficient and ideal natural product syntheses is just one example of how modern synthetic research is being geared towards striving to match nature’s own efficiency in this task. As such, the successful elaboration of a synthetic route that follows as close as possible the sequence of biogenetic bond-forming operations, known as biomimetic syntheses, are often incredibly efficient and
22
closer to ideality than abiotic variants. Very often a group will base their retrosynthetic planning on a rational analysis of the most probable biosynthetic route that was taken in reaching the target molecule.
1.3.3.2) Green chemistry and total synthesis
This notion of aiming for the ideal synthesis goes hand in hand with the other great challenge to total synthesis that has loomed on the horizon of the 21st century, that of producing ever more environmentally benign routes to the desired targets. This is part of a newly formalized perspective in chemistry, referred to as “green chemistry”, charged with the task of bringing chemical practices in line with the global effort to combat pollution, ecological destruction and climate change.57 Indeed, state-of-the-art organic synthesis has lagged behind other scientific pursuits in this regard. It has been roughly calculated that for every kilogram of fine chemical and pharmaceutical products produced using the most advanced and efficient synthetic techniques, up to 100 times that amount in chemical waste is regularly discarded.58 This “E” factor compares very unfavourably with other sectors of the chemical industry (see Table 1); realizations of this sort have contributed to a change in the criteria that chemists have traditionally relied upon to judge the merits of a total synthesis.
Industry sector Product tonnage kg byproduct/kg product
(E factor) Bulk chemicals 104-106 <1-5 Fine chemicals 102-104 5-50 Pharmaceuticals 10-103 25-100
TABLE 1: The “E” factor in the chemical industry58
The Environmental Protection Agency published its twelve principles of green chemistry as a way of guiding but also incentivising researchers to develop new technologies that are also green.59 Generally speaking these principles encourage the consideration of atom economy, safety, waste and energy reduction, greater reliance on catalysis and the incorporation of pollutant monitoring as part of a researcher’s objectives.57-60 Specifically for the evaluation of