Simulation of thermomechanical properties of U-PuO2 nuclear fuel under irradiation
155
0
0
Texte intégral
Figure

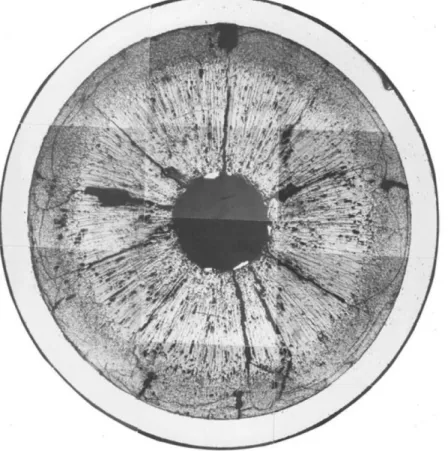
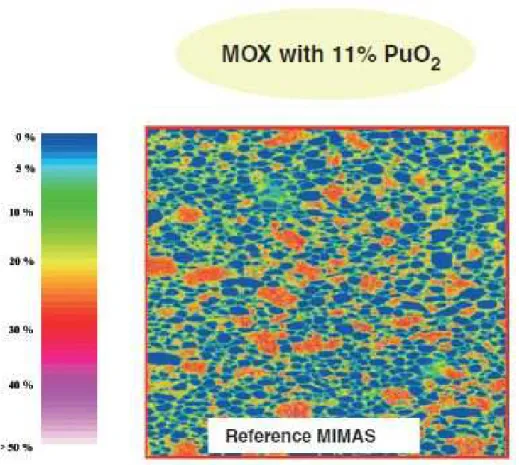
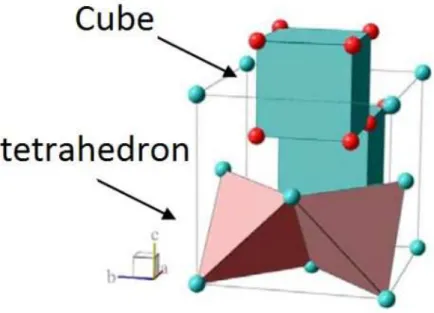
+7

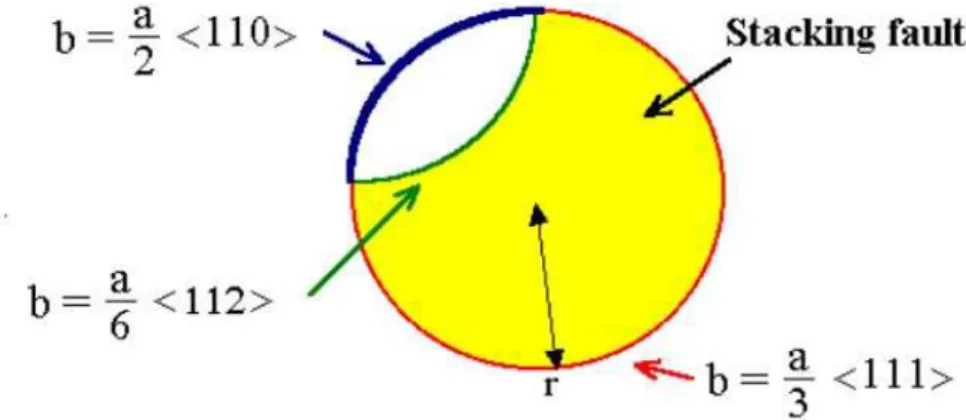
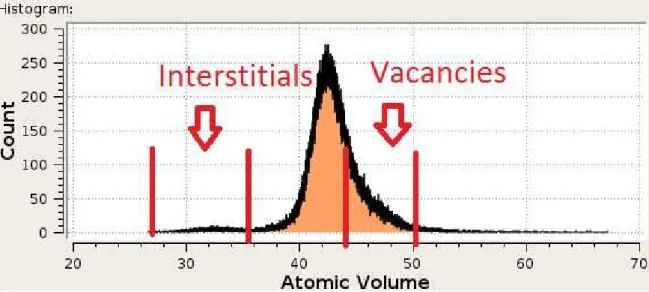
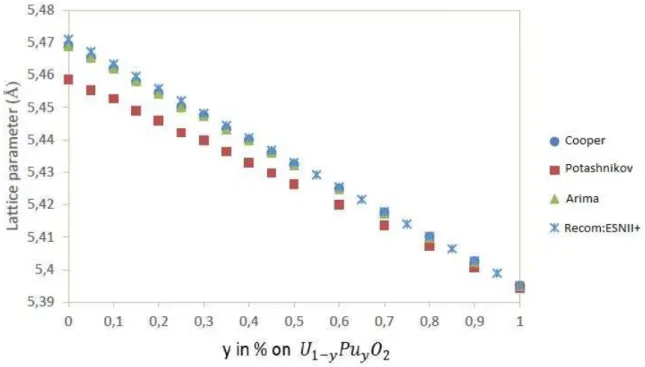
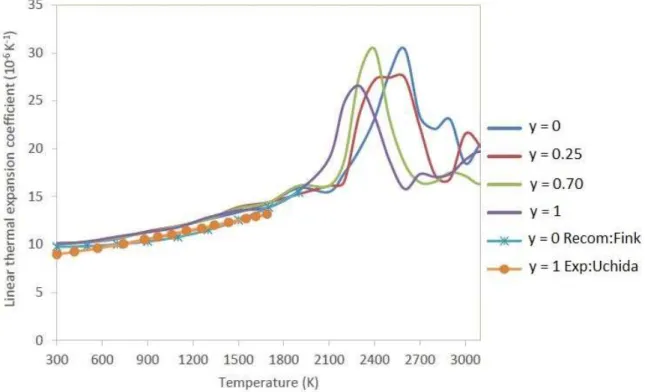

Documents relatifs