Trafic intranucléaire de l’ARN de la télomérase et la réponse aux dommages à l’ADN chez la levure Saccharomyces cerevisiae
Texte intégral
Figure




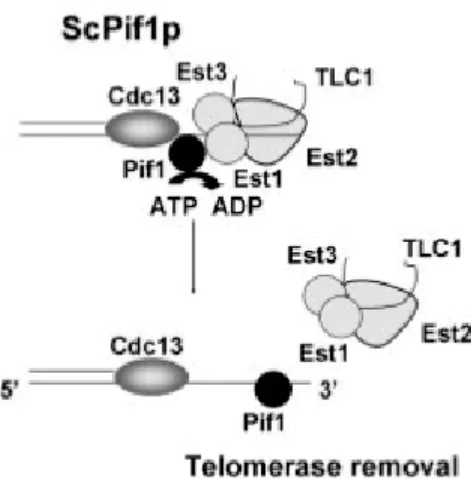
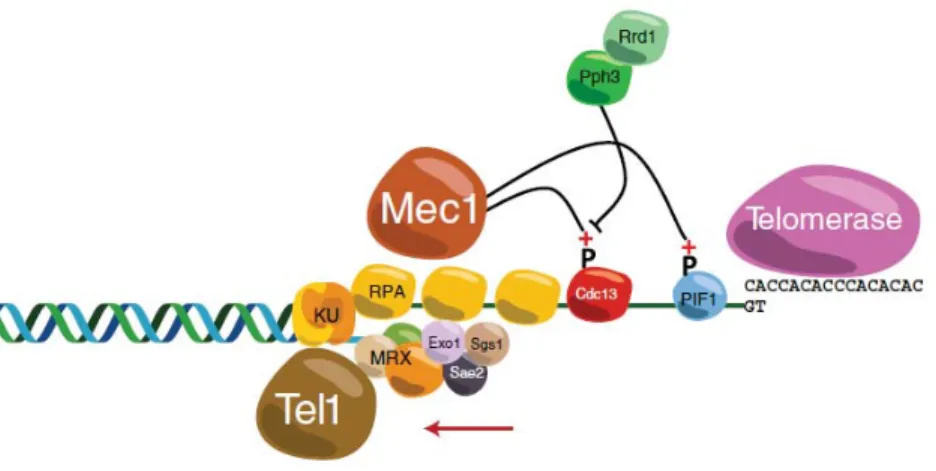
Documents relatifs
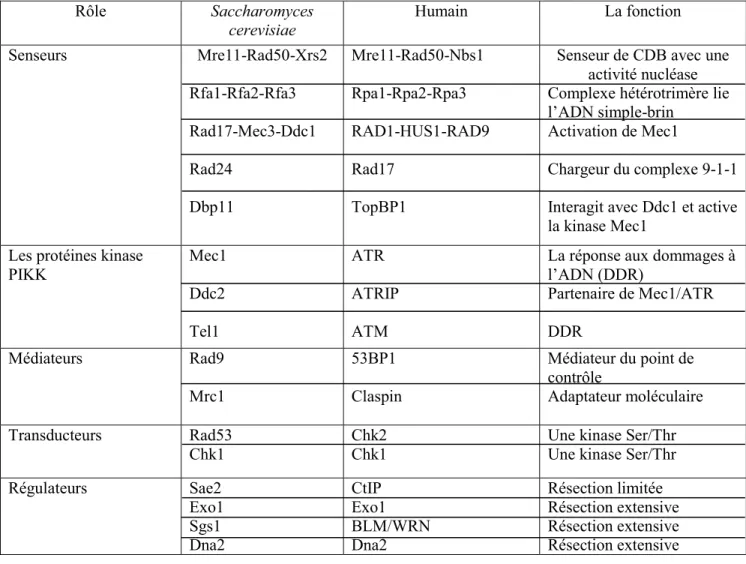
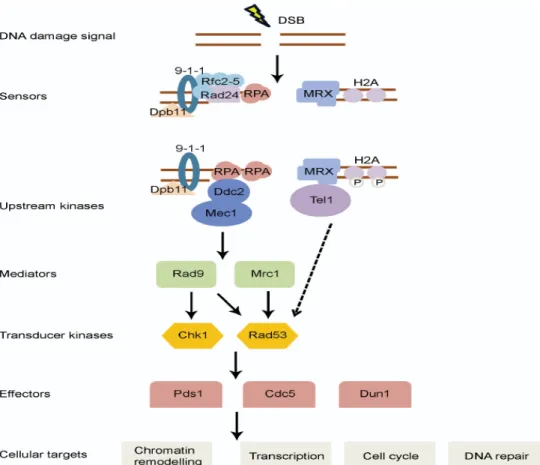
Les acteurs majeurs du checkpoint de phase S sont des protéines, décrites dans de nombreux mécanismes moléculaires, dont la gestion des dNTPs au cours du cycle
Enfin, les MPT (Shogren-Knaak et al., 2006) des histones et la présence in vivo de protéines courbant l’ADN (Bajpai et al., 2017) compromettraient l’établissement d’une structure
Comme les trois autres protéines de levure à domaine EH sont requises pour l’étape d’internalisation de l’endocytose, nous avons étudié cette voie de trafic dans des
Pour améliorer cette expérience , il est suggéré de réaliser des expériences avec la technique récente de test par ligature de proximité (PLA en anglais). Cela
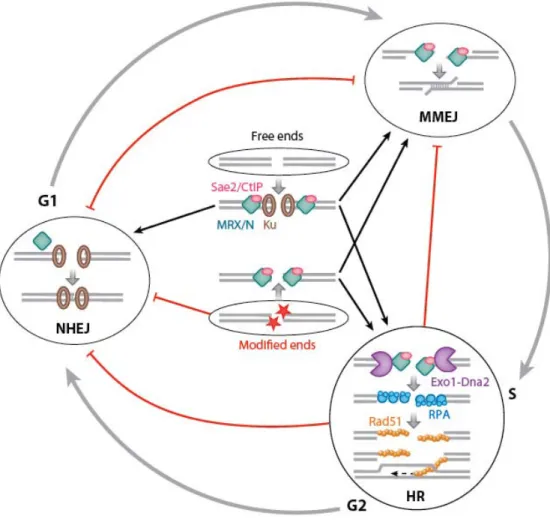
Afin de déterminer si les cellules cdc13Δ présentaient un phénotype d’adaptation, nous avons déterminé le taux de formation de ce type cellulaire en absence des gènes codant pour
Dégradation des substrats + ou non - verso Tableau 2 : Effet de la levure probiotique sur la dégradabilité ruminale de la MS % Tableau 3 : Influence d’un apport d’orge et de
En effet les gènes codant pour les sous-unités de l’exosome à ARN sont présents dans la majorité des génomes archées contrairement au gène codant pour aRNase J qui est
Enfin, les MPT (Shogren-Knaak et al., 2006) des histones et la présence in vivo de protéines courbant l’ADN (Bajpai et al., 2017) compromettraient l’établissement d’une structure
