The Independence of CXCR4’s Pathways, Gαi and
β-Arrestin2, and Their Modulation by AMD3100 and
TC14012
par Nassr Nama
Département de Microbiologie, Infectiologie et Immunologie Faculté de Médecine
Mémoire présenté à la Faculté de Médecine en vue de l’obtention du grade de maîtrise
en microbiologie et immunologie
Septembre, 2014
RÉSUMÉ
CXCR4 est un récepteur de chimiokines impliqué dans les métastases et la mobilisation des cellules souches hématopoïétiques. Il signale par deux voies: Gαi et arrestin2. La β-arrestine2 termine la signalisation des protéines G et cible le récepteur vers l'endocytose.
Un des objectifs du projet était d’étudier l’effet de certaines mutations de CXCR4 sur la signalisation et la localisation du récepteur. À l’aide de la technique BRET, nous avons confirmé que le mutant N119S est constitutivement actif sur la voie de signalisation Gαi. De plus, nous avons constaté que le mutant R134A était dépourvu de signalisation par les protéines G, mais qu’il recrutait constitutivement la β-arrestine2.
Nous souhaitions également étudier la dépendance du recrutement de la β-arrestine2 sur l’activité de Gαi. En utilisant la toxine de la coqueluche, un inhibiteur de la voie Gαi, le recrutement constitutif de la β-arrestine2 à R134A et N119S était maintenu. Ces résultats démontrent que, pour le récepteur CXCR4, le recrutement de la β-arrestine2 est indépendant de l’activation de Gαi.
Finalement, nous avons investigué la capacité de recrutement de la β-arrestine2 induite par deux ligands synthétiques de CXCR4, soit AMD3100 et TC14012. À noter, AMD3100 est un médicament déjà approuvé pour la transplantation des cellules souches. Par contre, il démontre des effets secondaires considérables. D’un côté, nous avons constaté qu’AMD3100 était un antagoniste sur les deux voies de signalisation, soit Gαi et β-arrestine2. De l’autre côté, TC14012 a démontré l’effet d’agoniste inverse sur la voie Gαi, mais d’antagoniste sur la voie β-arrestine2. À la lumière de ces résultats, TC14012 pourrait être plus approprié dans des cas cliniques puisqu’il réduirait toute activité basale de Gαi sans affecter le recrutement de la β-arrestine2. Enfin, ces résultats suggèrent que le ligand TC14012 pourrait être utilisé dans des essais cliniques visant la mobilisation des cellules souches.
Mots-clés: CXCR4, RCPG, AMD3100, Plerixafor, TC14012, CAM, CIM, motif DRY, N3.35
SUMMARY
CXCR4, a chemokine receptor involved in metastasis and homing of hematopoietic stem cells, signals through two major pathways: Gαi and β-arrestin2. β-arrestin2 terminates G-protein signaling and targets the receptor to endocytosis.
This project proposed to study the effect of a previously described set of CXCR4 mutants on both these signaling pathways, as well as their localization. These mutants were assayed by different Bioluminescence Resonance Energy Transfer (BRET) systems. Using these systems, we confirmed that N119S is a constitutively active mutant (CAM), spontaneously activating Gαi. As well, we found that R134A is a constitutively inactive mutant (CIM), devoided of G-protein signaling, but spontaneously recruiting β-arrestin2.
In addition, we studied the dependency of β-arrestin2 recruitment on the Gαi activity. By targeting R134A and N119S with pertussis toxin, an inhibitor of the Gαi activation, we showed efficient blocking of the Gαi pathway, while maintaining the constitutive recruitment of β-arrestin2. This demonstrated that for CXCR4, β-arrestin2 recruitment is independent of the Gαi pathway.
Finally, two synthetic ligands of CXCR4, AMD3100 and TC14012 were tested for their ability to recruit β-arrestin2. AMD3100 is a clinically approved drug used for stem cell transplantation, with considerable side effects. We found it to be an antagonist on both Gαi and β-arrestin2 recruitment. On the other hand, TC14012 was found to be an inverse agonist on Gαi and an antagonist on β-arrestin2 recruitment. Based on this finding, it would be preferable to use of TC14012 as it will further reduce any basal Gαi activity, without affecting β-arrestin2 recruitment. These results support the development of TC14012 for stem cell mobilization trials.
Keywords: CXCR4, GPCR, AMD3100, Plerixafor, TC14012, CAM, CIM, DRY motif, N3.35
ABBREVIATIONS
A Alanine
AC Adenylate Cyclase
ALL Acute Lymphoblastic Leukemia
AML Acute Myeloid Leukemia
AngII Angiotensin II
ANOVA Analysis Of Variance
AT1AR Angiotensin II type 1A Receptor
B.N.U BRET Net Unit
B.U BRET Unit
BRET Bioluminescence Resonance Energy Transfer C-terminal Carboxy-Terminal
Ca2+ Calcium ions
CAM Constitutively Active Mutant cAMP Cyclic Adenosine Monophosphate CCV Clathrin-Coated Vesicles
CI Confidence Interval
CIM Constitutively Inactive Mutant
D Aspartic acid
DNA Deoxyribonucleic Acid
EC50 Half maximal effective concentration
Epac Exchange protein directly activated by cAMP ERK Extracellular Signal Regulated Kinases FDA Food and Drug Administration
FRAP Fluorescence Recovery After Photobleaching G-CSF Granulocyte-Colony Stimulating Factor
GAP GTPase Activating Protein
GDP Guanosine Diphosphate
GEF Guanine Exchange Factor
GFP10 Green Fluorescent Protein 10 GPCR G-Protein Coupled Receptor
GRK GPCR Kinase
GTPγS Guanosine 5'-O-[Gamma-Thio] Triphosphate
HEK Human Embryonic Kidney cells
HSC Hematopoietic Stem Cell
K Lysine
M1 AChR Muscarinic acetylcholine receptor M1 MAPK Mitogen-Activated Protein Kinase mRFP Monomeric Red Fluorescent Protein
MVB Multivesicular Bodies
N Asparagine
N-terminal Amino-Terminal
PKC Protein Kinase C
PLC Phospholipase C
PTHR Parathyroid Hormone Receptor
PTX Pertussis Toxin
R Arginine
RGS Regulator of G-Protein Signaling Rluc3 Renilla Luciferase 3
S Serine
SDF-1α CXCL12, Stromal Cell Derived Factor-1α SEM Standard Error of the Mean
TM Transmembrane Helix
V1aR Vasopressin 1a receptor
V2R Vasopressin Type II Receptor VSMC Vascular Smooth Muscle Cell
wt Wildtype
Y Tyrosine
YFP Yellow Fluorescent Protein α1B-AR α1B-Adrenoceptor
α2A-AR α2A-Adrenoceptor
TABLE OF CONTENTS
RÉSUMÉ II
SUMMARY III
ABBREVIATIONS IV
TABLE OF CONTENTS VI
LIST OF TABLES VIII
LIST OF FIGURES IX
ACKNOWLEDGMENTS X
1. INTRODUCTION 1
1.1.G-PROTEIN COUPLED RECEPTORS (GPCRS) 1
1.2.SIGNALING 2
1.3.TRAFFICKING 6
1.4.CHEMOKINES 8
1.5.CHEMOKINE RECEPTORS 9
1.6.CXCR4 10
1.7.THE CONSERVED DRYMOTIF 15
1.8.THE ASPARAGINE N3.35 16
1.9.PHARMACOLOGY 17
1.10.AGONIST,ANTAGONIST AND INVERSE AGONIST 18
1.11.FUNCTIONAL SELECTIVITY 20
1.12.AMD3100 22
1.13.TC14012 24
2. OBJECTIVES 25
3. MATERIAL AND METHODS 26
3.1.MATERIALS AND PLASMIDS 26
3.2.CELL CULTURE AND TRANSFECTION 26
3.3. CAMPBIOSENSOR BRET2ASSAY 27
3.4.B-ARRESTIN2RECRUITMENT BRET2ASSAY 27
3.5.TITRATION ASSAYS 27
3.6.IMAGING OF CXCR4USING SPINNING DISC CONFOCAL MICROSCOPY 28
3.7.DATA ANALYSIS 28
4. RESULTS 33
4.1.GΑI ACTIVITY OF CXCR4MUTANTS 33
4.2.B-ARRESTIN2RECRUITMENT BY CXCR4MUTANTS 38
4.3.TITRATIONS OF B-ARRESTIN2RECRUITMENT 42
4.4. DIFFERENTIAL CONFORMATIONAL BASAL B-ARRESTIN2 RECRUITMENT BY CXCR4
MUTANTS 45
4.5.EFFECT OF PERTUSSIS TOXIN ON GΑI ACTIVITY AND B-ARRESTIN2RECRUITMENT 48
4.6.LOCALIZATION OF THE CXCR4MUTANTS 52
4.7.EFFECT OF AMD3100 AND TC14012 ON GΑI SIGNALING 56
4.8.EFFECT OF AMD3100 AND TC14012 ON B-ARRESTIN2RECRUITMENT 61
4.9.EFFECT OF TC14012 ON B-ARRESTIN2RECRUITMENT BY N119S 66
5. DISCUSSION 69
5.1.OVERVIEW 69
5.2.N3.35:N119 70
5.3.THE DRYMOTIF 71
5.4.B-ARRESTIN2RECRUITMENT MODALITIES 73
5.5.TRAFFICKING OF CXCR4MUTANTS 75
5.6.B-ARRESTIN2INDEPENDENT OF G-PROTEINS COUPLING 76
5.7.AMD3100 VS TC14012 77
6. CONCLUSION 79
7. PERSPECTIVES 80
LIST OF TABLES
TABLE I. GΑI ACTIVITY BY CXCR4 MUTANTS. 36
TABLE II. Β-ARRESTIN2 RECRUITMENT BY CXCR4 MUTANTS USING BRET2. 41 TABLE III. TITRATIONS OF Β-ARRESTIN2 RECRUITMENT BY CXCR4
MUTANTS. 44
TABLE IV. ANALYSIS OF TITRATIONS OF BASAL Β-ARRESTIN2
RECRUITMENT BY CXCR4 MUTANTS. 46
TABLE V. EFFECT OF PERTUSSIS TOXIN ON GΑI ACTIVITY. 50 TABLE VI. EFFECT OF PERTUSSIS TOXIN ON Β-ARRESTIN2 RECRUITMENT. 50 TABLE VII. EFFECT OF PERTUSSIS TOXIN ON Β-ARRESTIN2 RECRUITMENT.
51 TABLE VIII. EFFECT OF AMD3100 AND TC14012 ON GΑI ACTIVITY. 58
TABLE IX. EFFECT OF AMD3100 AND TC14012 ON Β-ARRESTIN2
RECRUITMENT. 64
TABLE X. TITRATIONS OF THE EFFECT OF TC14012 ON Β-ARRESTIN2
LIST OF FIGURES
FIGURE 1. SCHEMA OF CXCR4 AMINO ACIDS. 3
FIGURE 2. THE FOUR FAMILIES OF GΑ-PROTEINS. 4
FIGURE 3. TRAFFICKING OF GPCRS. 7
FIGURE 4. THE CXC CHEMOKINE FAMILY. 9
FIGURE 5. CXCR4 SIGNALING PATHWAYS. 11
FIGURE 6. HOMING OF HEMATOPOIETIC STEM CELLS. 13 FIGURE 7. IMPLICATION OF CXCR4 IN METASTASIS. 14
FIGURE 8. PHARMACOLOGY OF GPCR ACTIVITY. 17
FIGURE 9. AGONIST, ANTAGONIST AND INVERSE AGONIST. 18
FIGURE 10. AMD3100 23
FIGURE 11. TC14012 24
FIGURE 12. ASSAYING CAMP LEVELS USING EPAC REPORTER. 31
FIGURE 13. Β-ARRESTIN RECRUITMENT ASSAY. 31
FIGURE 14. ANALYSIS OF A TITRATION EXEPERIMENT. 32
FIGURE 15. GΑI ACTIVITY BY CXCR4 MUTANTS. 34
FIGURE 16. Β-ARRESTIN2 RECRUITMENT BY CXCR4 MUTANTS USING BRET2. 39 FIGURE 17. TITRATIONS OF Β-ARRESTIN2 RECRUITMENT BY CXCR4
MUTANTS USING BRET2. 43
FIGURE 18. TITRATIONS OF BASAL Β-ARRESTIN2 RECRUITMENT BY CXCR4
MUTANTS USING BRET2. 46
FIGURE 19. EFFECT OF PERTUSSIS TOXIN ON GΑI ACTIVITY AND
Β-ARRESTIN2 RECRUITMENT. 49
FIGURE 20. LOCALIZATION OF THE CXCR4 MUTANTS BY CONFOCAL
MICROSCOPY. 53
FIGURE 21. EFFECT OF AMD3100 AND TC14012 ON GΑI. 57
FIGURE 22. EFFECT OF AMD3100 AND TC14012 ON Β-ARRESTIN2
RECRUITMENT. 62
ACKNOWLEDGMENTS
I would like to thank Dr. Nikolaus Heveker for his constant enthusiasm to teaching, his motivational encouragement and his amazing support throughout the whole project.
As well, I want to thank all of the lab members; Nicolas Montpas and Stéphanie Gravel for their outstanding teaching and supervision, François Guité-Vinet for being a constant source of motivation, Guillaume Sylvain-Drolet for developing CXCR4 mutants, as well as Genevieve St-Onge, Marilou Lefrançois and Julien Bonneterre.
Also, I would like to express my appreciation to Dr. Gilles Hickson and his laboratory. They have offered me the possibility to work with them and learn from their microscopy expertise.
Thank you to Tyler James, Thinh Nguyen and Richard Hae for proofreading this thesis.
Finally, a special thanks to the Mach-Gaensslen Foundation of Canada and the Canadian Institutes for Health Research for funding this study.
1. INTRODUCTION
1.1. G-protein Coupled Receptors (GPCRs)
The seven-transmembrane domain receptors, more commonly known as G-protein coupled receptors (GPCRs), are the largest family of integral membrane proteins on the cell surface (1), and are found only in eukaryotic cells (2). This family of receptors modulates several important physiological responses, such as cardiovascular and renal functions, as well as neurotransmission. With their central implication in the functioning of several systems, there is no surprise in the significance of their deregulation (3). The importance of these receptors is demonstrated by the fact that 30–40% of prescribed drugs target this family (4). The ligands of these receptors are diverse ranging from amino acids, proteins, hormones, peptides, nucleotides to odorant molecules and even some ions (1).
The name of these receptors comes from their structure, consisting of seven transmembrane alpha helices (TMs). These hydrophobic structures are separated by three intracellular loops and three extracellular ones (Fig. 1) (5). The extracellular surface is composed of the N-terminal tail with the extracellular loops, which forms the binding surface of the receptors’ ligands. On the other hand, the three intracellular loops and the C-terminus form the site of interaction with the proteins that regulate the activity of the receptors, such as the G-proteins.
Classes: The GPCRs are stratified according to the GRAFS classification system (6). This acronym stands for the five families of receptors: Glutamate, Rhodopsin, Adhesion, Frizzles/taste2, and Secretin.
The chief interest of this project is a chemokine receptor, CXCR4, belonging to the rhodopsin-like family. This family will, therefore, be discussed in more detail. It is the largest family of receptors containing almost 700 members. Most of these show a conserved E/DRY motif in the second intracellular loop, next to third transmembrane helix (TM III) (Fig. 1). Among the members of this family, we distinguish the adrenergic, the chemokine, and the opioid receptors (6).
1.2. Signaling
The GPCRs exert their signaling mainly by coupling with the heterotrimeric G protein family (7). However, it is clear now that the GPCRs can signal through mechanisms
independent of the G proteins, mainly through beta-arrestins (β-arrestins).
G-proteins: The heterotrimeric G proteins are the main intracellular effectors associated with GPCRs. They belong to the family of proteins called GTPases and are composed of three distinct proteins called alpha, beta, and gamma (α, β, and γ) (1). In its inactive form, the Gα subunit is bound to a guanosine diphosphate (GDP), thus enabling the Gα subunit to bind noncovalently to the Gβγ complex.
The binding of a GPCR ligand induces conformational changes within this receptor, leading to the activation of the heterotrimeric G proteins. The activated receptor plays the role of a guanine exchange factor (GEF), which means that it induces the exchange of the GDP attached to the α subunit with a guanosine triphosphate (GTP) (1,8). The activated G-proteins lead to the subsequent activation of the downstream effectors. The Gαsubunit has an intrinsic GTPase activity, leading to the eventual hydrolysis of GTP to GDP and the termination of the signaling process. This GTPase activity could be accelerated by a GTPase-accelerating protein (GAP), such as the regulators of G-protein signaling (RGS) (1,1,8,9).
Sixteen Gα genes have been identified so far, and alternative splicing can generate twenty Gα proteins. On the basis of their sequence, they can be grouped into four families: Gαs, Gαi/o, Gαq, and Gα12/13 (Fig. 2) (1). The Gαs family regroups the different Gα subunits that stimulate adenylate cyclase (AC), while the Gαi/o family contains the Gα subunits capable of inhibiting this enzyme, Gαi and Gαo. The Gαq family includes those that activate phospholipase C (PLC), such as Gαq and Gα11. Finally, the Gα12/13 family regulates the cytoskeletal assembly through its two effectors Gα12 and Gα13 (10).
Previously the general assumption was that each GPCR is coupled to one type of Gα subunit. However, it is now widely accepted that it could be coupled with different ones, depending on the tissue expression and the cellular localization (11).
Figure 1. Schema of CXCR4 amino acids. CXCR4 receptor portrayed as a chain of amino acids. Seven transmembrane helices, three intracellular loops, three extracellular loops, an N-terminal and a C-N-terminal compose the receptor. The DRY motif and the conserved Asparagine N119 are highlighted in red. The residues highlighted in black are important for ligand binding and signaling. Modified from Doranz 1999 (8).
G protein-coupled receptor kinases (GRKs): Following stimulation, the GPCRs are phosphorylated by GPCR kinases (12). The GRKs 1-7 phosphorylate residues on the C-terminal of the GPCRs or the third intracellular loop. This phosphorylation is one of the mechanisms used to terminate G-protein signaling. However, its role is more complex as it induces a second wave of signaling independently of the G-proteins (13). The roles of these different GRKs are not purely redundant. Different GRKs exhibit different functions, by phosphorylating different residues of the GPCR. For instance, GRK5/6 activates β-arrestin
signaling, associated with vasopressin type II receptor (V2R) and β2-adrenoceptors (β2-AR)
(14-16). On the other hand, phosphorylation by GRK2/3 negatively regulates this signaling pathway.
Figure 2. The four families of Gα-proteins. The Gαs, and the Gαi stimulates or inhibits the Adenylate Cyclase (AC) respectively, thus affecting the levels of cAMP. Gαq activates Phospholipase C (PLC), leading to the activation of Protein Kinase C (PKC), and Ca2+ release. Gα12/13 activates RhoGEF, leading to the activation of RhoA. Finally, the activated complex Gβγ activates Raf, and the ERK1/2 pathway. Modified from Zhang 2012 (17).
β-arrestin: β-arrestin proteins (1 and 2) bind to the phosphorylated receptors and shut off signal transduction in a process called desensitization. They then target the desensitized receptor to clathrin-coated pits and endocytosis. Recruitment of β-arrestin to the activated receptor allows assembly of the endocytic machinery and targeting of the receptor to the clathrin-coated vesicles (CCVs) (18). At the same time, β-arrestin functions as a scaffolding protein by recruiting several interacting proteins such as E3 ubiquitin ligases or Mitogen-Activated Protein Kinases (MAPKs) (19,20).
The two isoforms of the β-arrestin share a common sequence of 78% (21). Even if the functions of the two β-arrestins are often interchangeable, the presence of at least one of them is crucial, as knocking out both is lethal in animal models (22). Nevertheless, they are not always functionally redundant. For example, the internalization of the β2-Adrenoreceptor (β-2AR) is dependent on β-arrestin2, while that of proteinase-activated receptor 1 (PAR1)
depends on β-arrestin1. The preferential association of a GPCR to one of the two β-arrestins depends on a GPCR classification that is discussed later, in the ‘1.3. Trafficking’ section. According to this classification system, Class A receptors show a preference for β-arrestin2 over β-arrestin1, while class B receptors have equivalent affinities for both (18).
The recruitment modalities of β-arrestin include the affinity between the two proteins, the distance between the two, the conformation of the interaction, as well as kinetics along with the dynamics of the binding. Recruitment of β-arrestin can occur in various conformations (23). β-arrestin acts as a scaffolding protein and recruits different proteins depending on its conformation. While some conformations may be more compatible with the recruitment of the endocytic machinery, others may be more efficient to activate the MAPKs. This shows that even the recruitment of β-arrestin can be regulated, leading to the activation of various signaling pathways. The recruitment of β-arrestins is dependent on two factors, the change in the receptor conformation induced by the agonist and the phosphorylation of specific sites in the receptor’s C-terminus (24). The differential modalities observed here might be due to either of these two factors. The phosphorylation of the C-terminus generates a barcode that is different, depending on the phosphorylation sites. This differential phosphorylation leads to recruitment of β-arrestins in various conformations, thus affecting its biological functions (24).
Mitogen-activated protein kinase (MAPK): Several pathways are recruited by β-arrestins and MAPK is the best understood one. The MAPK family includes several members, such as the extracellular-signal-regulated kinases (ERK) 1 and 2. These pathways have an impact on target gene transcription, cell-cycle progression, and apoptosis (22,25). The ERK1/2 pathway consists of MAPK kinase kinases (Raf), MAPK kinases (MEK1/2), and finally the MAPKs (ERK1/2). As a scaffolding protein, β-arrestin ensures an efficient arrangement of the complex of these three proteins (18). Thus, activation of the GPCR by its ligand leads to the recruitment of β-arrestin, and also the following activation of the preformed complex Raf-MEK-ERK on this protein (25,26). This activation requires that β-arrestin is in a conformation capable of recruiting these proteins, and their subsequent activation. As β-arrestin can be recruited in various conformations, not all are compatible with the activation of the MAPK.
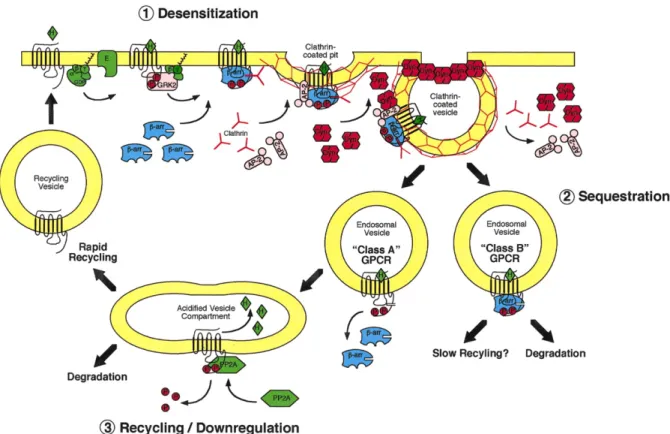
1.3. Trafficking
Endocytosis of the GPCRs is a complex process. It starts mainly with the phosphorylation by GRKs, the recruitment of β-arrestins and is followed by clathrin-mediated endocytosis. Although this process was first regarded as a means to desensitize the stimulated receptor, it is now clear that it involves G-independent signaling pathways.
As mentioned earlier, the phosphorylation of the GPCR by a GRK and the recruitment of β-arrestin to the activated receptor allow the assembly of the endocytic machinery and the internalization of the complex in the clathrin-coated vesicles (CCVs) (19). These vesicles proceed to form endosomes containing the activated receptor bound to β-arrestin. Once in these endosomes, the GPCRs can either recycle to the plasma membrane or be directed for degradation in the lysosomes (27). Depending on the stability of the interaction of GPCR-β-arrestin, two patterns of trafficking can be observed, and the GPCRs can be organized in two classes (A or B) (Fig. 3) (18,22).
First, the Class A receptors show a high affinity for β-arrestin2 compared to β-arrestin1 (28). They interact transiently with β-arrestin2 and form complexes that dissociate quickly after being internalized (29). This rapid dissociation of β-arrestin permits the receptor to recycle to the cell membrane. This is the case for the β2-AR, which upon stimulation with its endogenous ligand is rapidly desensitized, dephosphorylated, and recycled to the cellular surface (18,30).
In contrast, the class B receptors have a stronger interaction with β-arrestin, leading to a slower desensitization process (18). The receptors and their β-arrestins form a strong complex that can even be observed on the endosomes. This higher affinity slows down receptor recycling and directs the majority of the receptors toward degradation.
Ubiquitination: The trafficking and sorting of the GPCRs after endocytosis is closely dependent on their ubiquitination (31). β-arrestin2 plays an important role in this ubiquitination, as it is likely to function as an adaptor for E3 ligase, the third enzyme of the ubiquitination process (32,33). This ubiquitination will determine the receptor’s final fate,
degradation or membrane recycling. Monoubiquitination of a receptor is associated with its internalization (34), while its polyubiquitination acts as a lysosomal sorting signal (35,36). The CXCR4 receptor follows this pattern of ubiquitination. Lysine residues on the C-terminal of the receptor are ubiquitinated and the receptor is directed to degradation (37).
Figure 3. Trafficking of GPCRs. After activation of the GPCR, the GPCR kinase (GRK) phosphorylates the C-terminal of the receptor, leading to the recruitment of β-arrestin, and the endocytic machinery. After endocytosis, receptors follow one of two patterns, class A receptors lose the β-arrestin and are either rapidly recycled or degraded, while class B receptors are mainly directed to degradation. Modified from Luttrell 2002 (38).
1.4. Chemokines
Chemokines are a family of cytokines displaying a chemotactic activity and orienting the cells throughout the organism. They are small basic proteins consisting of 70 to 130 amino acids, varying between 8kDa and 12kDa in size (39,40). Chemokines exhibit their main functions by signaling through GPCRs. Initially, chemokines had been associated with inflammatory responses, given their role in leukocyte migration (41). Furthermore, some are equally involved in cellular survival and growth (42). On the basis of their function, chemokines can be divided into two groups: constitutive chemokines and inflammatory ones. The former group comprises chemokines constitutively expressed and involved in the development and homing of stem cells. The latter group contains proinflammatory cytokines, whose expression is induced during an inflammatory response (41).
The diversity of the functional roles of chemokines makes their receptors attractive therapeutic targets (41). The Food and Drug Administration (FDA) has approved two such therapeutics drugs, Maraviroc, and Mozobil, targeting CCR5 and CXCR4, respectively (25). The major problems facing the development of new therapeutic molecules are the promiscuity and the redundancy of chemokines (40,41). Promiscuity is due to the binding of one chemokine to more than one receptor, while redundancy is related to the similar physiological functions exhibited by various chemokines (40).
More than 50 different chemokines have been identified so far, and they are organized in four different classes (Fig. 4) (40). This system of classification depends on a conserved two-cysteine motif in their N-terminal: CXC, CC, C, and CX3C, where the letter X denotes any amino acid. On the basis of this system of classification, each chemokine is identified by the class to which it belongs, the letter ‘L’ for the ligand, and a number based on their chronological order of discovery. For example, the chemokine SDF-1 belongs to the family CXC and, therefore, it is identified as CXCL12. The receptor’s nomenclature follows the chemokine it binds to, by adding the letter ‘R’ for receptor and a number that is not necessarily related to the chemokine number (43). Despite the introduction of this classification system, the old nomenclature is widely used (44).
Figure 4. The CXC chemokine family. Chemokines are classified depending on four conserved cysteines, into four families CXC, CC, C, and CX3C. X represents any amino acid. Modified from Townson 2003 (45).
1.5. Chemokine Receptors
Chemokine receptors are GPCRs (46). Twenty receptors belonging to this family have been identified (40). They are involved in the body’s homeostasis, as well as in the coordination of the two branches of the immune response — the innate and acquired immune responses. Deregulation of proper chemokine functions leads to serious immunological problems. This results in an inefficient immune system either failing to respond adequately to infections or becoming overly responsive to exogenous substances, as in asthma, or one’s own-self, as in rheumatoid arthritis (RA) (41).
Chemokine receptors belong to the Rhodopsin-like family. They share 25–80% of their amino acid sequence identity, as well as several other characteristics (40). Indeed, chemokine receptors have a highly conserved DRY motif in the second intracellular loop, and a cysteine residue in each extracellular loop, enabling them to form disulfide bridges, ensuring the proper folding of the receptor and stabilizing the surface of interaction with the ligand (47).
The expression of chemokine receptors is not specific to immune cells. Chemokine receptors are also found on epithelial cells, endothelial cells, neurons and glial cells (40).
1.6. CXCR4
CXCR4 is a GPCR (48), which binds one endogenous chemokine ligand, CXCL12 (Fig. 1). CXCL12 is a constitutive chemokine, implicated in cellular migration during various stages of development, such as organogenesis and hematopoiesis (48,49). During adult life, CXCR4 plays a pivotal role in hematopoietic stem cell homing and their retention in the bone marrow (50). CXCR4 is equally involved in different pathologies including tumors, metastasis, and inflammatory diseases (49). As well, it serves as a secondary co-receptor for some strains of HIV (51). Therefore, we can assume that CXCR4 is a valid potential target for several pathologies.
CXCR4 has only one physiological ligand, CXCL12. CXCL12 is a peptide of 68 amino-acids that assumes a tertiary structure of a compact core and a flexible N-terminus (52). CXCL12 binds to the external domains of CXCR4 (53). This binding leads to conformational changes in CXCR4, and thus, its activation (52-55). After activation by CXCL12, CXCR4 signals through two major pathways: Gαiand β-arrestin (Fig.5)
1) G-proteins:Upon agonist binding, CXCR4 activates Gαi(56). Gαi inhibits AC, which is responsible for generating cyclic adenosine monophosphate (cAMP). The β/γ complex on the other hand activates PLC-β, Phosphatidylinositol 3’-Kinase (PI3K) and Protein Kinase C (PKC). These pathways result in a cytosolic Ca2+ influx and activation of the MAPK (48), leading to regulation of gene transcription and cell migration. In addition, CXCR4 activates small G-proteins such as Rho, Rac, and Cdc42, the central regulators of actin polymerization and chemotaxis (57). This signaling pathway is sensitive to the pertussis toxin (PTX). This toxin inhibits Gαi signaling by ADP-ribosylating the α subunit (58). Although Gαi is the main G-protein activated by CXCR4, it is not the only one. Several reports have identified Gαq/11 (59-61), and Gα12/13 (60,62) as pathways activated by CXCR4 and both are necessary for chemotaxis.
2) β-arrestin: After stimulation with CXCL12, CXCR4 is phosphorylated by GPCR kinases (GRKs), such as GRK2, 3, 5, and 6 (12). The β-arrestin proteins (1 and 2) bind to the phosphorylated receptors and shut off signal transduction in a process called
desensitization (12). β-arrestins target the desensitized receptor to clathrin-coated pits and endocytosis. Following internalization, CXCR4 may be slightly recycled back to the membrane, but it is mainly targeted for degradation by the lysosomes. β-arrestins are also able to induce signaling effects by activating the ERK pathway leading to enhanced chemotaxis and cellular survival (63).
As mentioned earlier, the roles of various GRKs are not merely redundant. For CXCR4, phosphorylation by GRK2 and 6 leads to the recruitment of β-arrestin1 and the termination of Gαi signaling, while phosphorylation by GRK3 and 6 induces the recruitment of β-arrestin2 and the subsequent activation of the ERK pathway (14).
Figure 5. CXCR4 signaling pathways. Activation of CXCR4 by its ligand CXCL12 leads to the activation of Gαi, and β-arrestin pathways. These lead to the activation of the different proteins illustrated in the figure. Figure taken from Teicher 2010 (64).
Physiological functions of CXCR4: The CXCL12–CXCR4 axis is functionally prominent. As mentioned earlier, it is implicated in hematopoiesis and in the immune response. The importance of this axis is demonstrated in the knockout animal models. Mice lacking the two genetic copies of either CXCR4 or CXCL12 die perinatally due to defects in the development of the heart and brain, GI tract vascularization, hematopoiesis and B-cell lymphopoiesis (1).
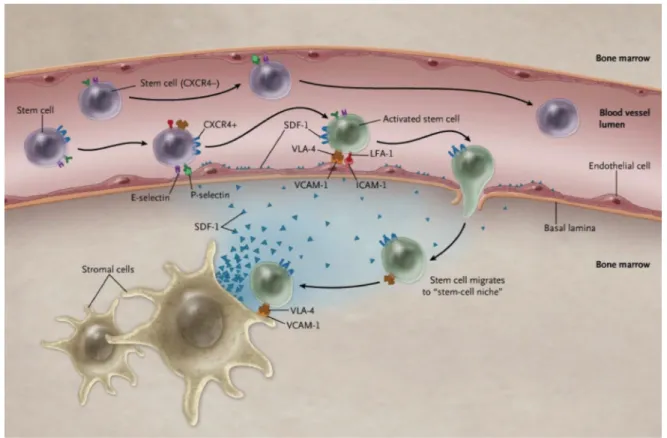
Mobilization and homing of hematopoietic stem cells: Homing is the process of migration of hematopoietic stem cells (HSCs) through the blood stream, toward various organs or the bone marrow (Fig. 6). In the opposite process, mobilization involves the recruitment of HSCs from the marrow to the blood stream (50,65). Both processes are modulated through CXCL12 and its receptor CXCR4 (66). Signaling through CXCR4 induces the homing of the HSCs and their retention in the bone marrow (67,68), while the interruption of this pathway leads to the mobilization of HSCs into the bloodstream (50). In the bone marrow, HSCs play an active role by self-renewing and having the potential to repopulate the blood cells (67,69). These HSCs are preserved in a microenvironment (niche) composed of stromal cells secreting CXCL12 (70). The commonly used name for this chemokine is SDF-1, which stands for the Stromal Cell-Derived Factor (69). Since this chemokine is secreted in the bone marrow niches, they contain its highest concentration. Diffusion of CXCL12 leads to the generation of a gradient capable of attracting the HSCs to populate these niches. Even though the low concentration of CXCL12 in the periphery promotes proliferation and migration of cells, the high doses present in the bone marrow niches induce cellular survival and quiescence (71).
Cancer and Metastasis: CXCR4 is the most widely expressed chemokine receptor on cancer cells (72), where it is highly expressed on several tumors of the breast, prostate, ovaries, and melanoma. This level of expression is correlated with tumor malignancy or its ability to metastasize (73). CXCR4 signaling induces proliferation and cell survival in some types of tumors (72). Also, the receptor has been proposed as a prognostic marker, and its inhibition by gene targeting or antibody blocking has stopped tumor growth and metastasis in several preclinical animal models.
Figure 6. Homing of hematopoietic stem cells. Stem cells in the blood stream are expressing CXCR4 (blue receptor). CXCL12 (SDF-1) is expressed by the stromal cells of the bone marrow, released, diffuses from its source and is present on the surface of the endothelial cells. Activation of stem cells by CXCL12 leads to their direction to the bone marrow. Different integrins play important roles in the interaction between the stem cells and the endothelial wall. Figure taken from Homan 2011 (74).
Metastasis is the major cause of mortality in cancer (75). It arises as a spread of the primary tumor and formation of secondary metastatic tumors. Detection of cancer in patients early, before the metastatic process, allows for an increased chance of treatment by chemotherapy, radiotherapy or surgery (76). As the tumor spreads throughout the body, the prognosis of the patient decreases dramatically, due to the difficulty of controlling and treating all the cancerous foyers. Therefore, it is primordial to understand the process of the metastatic spread and the mechanisms that contribute to it, to be able to predict its targets and to develop specific drugs to obstruct it.
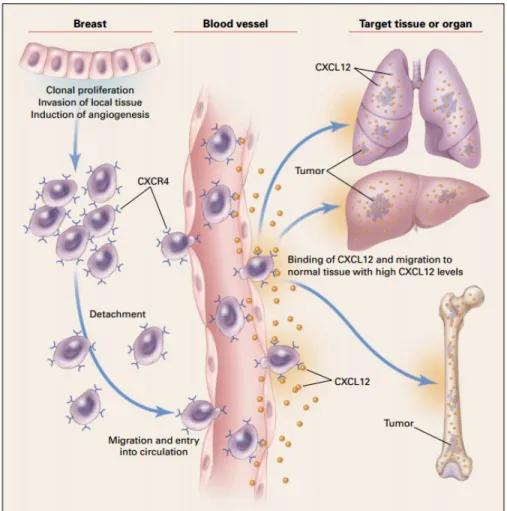
Metastatic dissemination is extremely regulated in the process of cancer spread. Metastatic cells tend to co-opt chemokine signals that are usually used for leukocyte trafficking (73). While CXCR4 is significant in primary tumor development, it is also essential in directing the migration of cancer cells invading the secondary organs (48,73). Activation of CXCR4 leads to the stimulation of signaling pathways required for migration (77). In addition, CXCL12 dictates the profile of metastasis in the CXCR4 expressing breast cancers (Fig. 7) (73). For example, the cancerous cells that express CXCR4 tend to migrate to organs with a high level of expression of this receptor’s ligand, CXCL12 (73), such as the lungs, the liver and the bone marrow (78).
Figure 7. Implication of CXCR4 in metastasis. Cancer cells from the primary breast tumor expressing CXCR4 lose attachment and enter the circulation. In the blood vessels CXCR4 interacts with CXCL12 present on the endothelial cell surface leading to their exit from the circulation and migration towards the organs expressing high levels of CXCL12, such as lungs, liver, and bone marrow. Figure taken from Murphy 2001 (75).
1.7. The Conserved DRY Motif
The DRY-motif is situated in TMIII, at the 3.49 position (Fig. 1). This is according to the Ballesteros–Weinstein nomenclature, where the first number reflects the transmembrane helix and the most conserved residue in this helix is given the number 50 (79). The DRY-motif is the most conserved motif in GPCRs and it has been reported to interact directly with the Gα proteins (80). To highlight the importance of this motif, Arginine 3.50 is conserved among 100% of the chemokine receptors. As well, in 95% of these GPCRs, the position 3.51 is occupied by an aromatic residue and a negatively charged residue at 3.49 (81). In the inactive form, the aspartic acid residue (D 3.49) and the arginine one (R 3.50) interact together (79). This interaction is interrupted during activation, leading to the release of arginine and its interaction with the Gα proteins. The importance of R3.50 in the activation and functioning of the chemokine receptors has been demonstrated after studying the DRY motif mutants (82). This residue has been revealed to be primordial to the G-protein signaling of several receptors, such as Muscarinic acetylcholine receptor M1 (M1 AChR) and Vasopressin 1a receptor (V1aR)
(82). In these receptors, as well as in CXCR4, the arginine of the DRY motif is involved in the direct coupling of the receptor to the G-proteins and the conformational changes induced by their respective agonists.
Mutating residues of the DRY-motif leads to two types of functionally variant mutants, the constitutively active ones (CAMs) and the constitutively inactive ones (CIMs). The CAMs spontaneously activate G-protein signaling (82,83), while the CIMs constitutively recruit β-arrestin and are deprived of G-protein signaling. Today, CXCR4 CAMs have been identified, but no CIM has yet been described.
Berchiche et al. had previously studied the DRY mutants for CXCR4 (84). Mutating the D3.49 to asparagine (D133N) or the Y3.51 to alanine (Y135A) gave a signaling pattern similar to that of the wild-type receptor. Arginine mutation (R134A) led to a receptor unable to activate the G-proteins. Nevertheless, it was not clear whether this mutant could demonstrate any signaling activity at all. The expression of these mutants on the cell surface was found comparable to the wild-type receptor (84). Both D133N and R134A had a slight decrease in
cell surface expression, which could be explained by several mechanisms: decrease in receptor’s stability, increase in internalization, or even a decrease in the receptor’s delivery to the cell surface by the trafficking machinery.
While these mutants have been tested with several techniques, neither the Gαi pathway nor β-arrestin2 recruitment have been studied specifically. The previous two reports studying this motif in CXCR4 focused on the recruitment of guanosine 5'-O-[gamma-thio] triphosphate (GTPγS), calcium influx, and gene transcription, all of which are shared among the G-proteins, without being specifically related to the Gαi subunit.
1.8. The Asparagine N3.35
The asparagine residue N3.35 is conserved among chemokine receptors (25). This residue is essential in the conformational changes induced during the activation of these receptors (85). Zhang et al. studied this amino acid in the chemokine receptor CXCR4 (85). Changing asparagine N119 to alanine or serine generated CAMs on the G-protein pathway. In this study, both mutants were able to constitutively activate a galactose gene reporter, recruit GTPγS to the plasmic membrane, induce a calcium influx and confer a constitutive phosphorylation to the C-terminus of the receptor (84,85). Furthermore, this constitutive activity was sensitive to pertussis toxin (PTX) indicating that it was associated with the Gαi pathway. Other mutations had different signaling patterns. The mutant N119D showed a similar activity to the wild-type in all three pathways mentioned above, which were related to the G-proteins. Surprisingly, the mutant N119K had no G-protein signaling. Nevertheless, all these mutants were adequately expressed on the cell surface (84), and their binding to the CXCL12 did not vary significantly (85).
Similar to what was mentioned for the DRY motif, this residue has not been studied for its role in specifically recruiting β-arrestin2.
1.9. Pharmacology
Several pharmacological parameters are used in this thesis and are, therefore, introduced here.
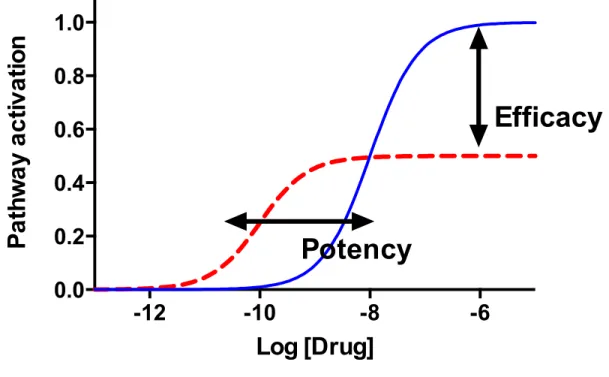
Figure 8. Pharmacology of GPCR activity. Schematic representation of the activity on one pathway as a function of the drug’s concentration. The concentration is presented on a logarithmic scale. Potency represents the concentration of a drug required to induce a response while efficacy represents the maximal response induced.
Affinity: The affinity describes the strength of interaction between the ligand and its receptor or two proteins (86). This strength depends on the chemical forces resulting in the interaction. Several types of forces stabilize this interaction, such as electrostatic, hydrophobic, hydrogen bonds or Van der Waals. This affinity can be measured by radio-binding assays. Here, the study focuses on the effect of different ligands on the relative affinity between CXCR4 and β-arrestin2, and a BRET titration assay is used for that. Although this type of assay does not provide quantifiable data on the affinity of the interaction, it permits comparison of the affinity among different conditions.
-12
-10
-8
-6
0.0
0.2
0.4
0.6
0.8
1.0
Log [Drug]
Pa
th
w
a
y
a
c
tiv
a
tio
n
Potency
Efficacy
Efficacy: Efficacy is a measure of the capacity of a ligand to induce a physiological effect (Fig. 8). It represents the maximal response that this ligand can induce on a pathway. Efficacy is measured using dose-response sigmoidal curves. With saturating concentrations of the ligand, the observed effect attains a plateau that represents the efficacy of this ligand (86).
Potency: Potency represents the concentration of a ligand required to induce a response (86). It is usually measured as EC50, the concentration of the ligand required to produce 50% of the maximal response (the efficacy). The lower the EC50 is, the more potent the ligand, meaning that even a small concentration of it can induce a response.
1.10. Agonist, Antagonist and Inverse Agonist
A receptor’s ligand is defined as a molecule with an affinity for the receptor. Still, its effect on receptor signaling can vary. Endogenous or synthetic ligands of a GPCR are classified as agonists, antagonists, or inverse agonists.
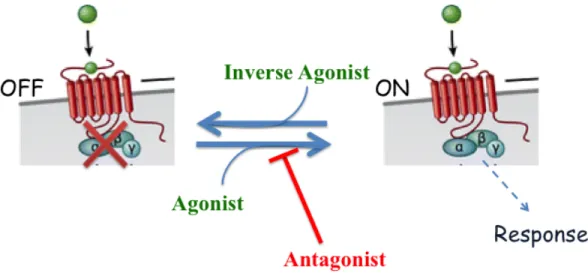
Figure 9. Agonist, antagonist and inverse agonist. A receptor represented as it has two states, an inactive (off) state, and an active (on) one giving a response. The equilibrium between the two is affected by the type of the ligand bound to the receptor. An agonist drives the equilibrium towards the active form; the inverse agonist changes it in the opposite way, while an antagonist blocks the receptor in its basal state. Modified from Zhang 2012 (17).
Agonist: An agonist is a ligand capable of inducing a response and also increasing the basal signaling of the receptor (Fig. 9) (87). It favors a transition to the activated conformation of a receptor, in respect to a particular pathway. The degree of agonism depends on the ligand and it can vary between a full agonist effect with a maximal response or a partial one. Often, endogenous ligands are those that show a maximal response. This difference in efficacy of agonism does not depend on the concentration of the ligand, as receptors are saturated with either ligand when measuring its efficacy. Rather, it is the intrinsic capacity of the ligand that dictates its maximal response.
Antagonist: An antagonist is a ligand capable of binding the receptor, and causing a block in its conformation without activating a signaling pathway (86,87). As it has no intrinsic activity, its effect can be observed when combined with an agonist. In this context, the antagonist would decrease the effect induced by the agonist on a particular pathway, either by competing with this latter for the binding site (Orthosteric Antagonist) or by allosterically modifying its activity (Allosteric Antagonist). An antagonist may or may not be competitive. A competitive antagonist sterically competes with the agonist and, by increasing the agonist’s concentration, can rescue its activity on this pathway. However, with a non-competitive antagonist, such as an allosteric one, adding increasing amounts of the agonist will not be able to counteract the antagonist effect.
Inverse Agonist: Finally, a ligand can also act as an inverse agonist. The inverse agonists reduce the basal signaling of a receptor in respect to a signaling pathway (88). Observing this activity requires a receptor with a basal constitutive activity (CAM) on this pathway, in the absence of any ligand. Stimulating these receptors with an inverse agonist demonstrates its negative efficacy (89,90). In the absence of such a constitutive activity, the inverse agonist acts as a mere antagonist.
1.11. Functional Selectivity
Biased signaling: A central aspect in the pharmacology of GPCRs is the concept of biased signaling (91), where a receptor state is not merely on or off, but the on/off must be used in terms of a particular pathway. A receptor’s ligand may be an agonist on one pathway, but not on the other (92).
Previously, the activation of the receptor has been explained by using the two-state model. This theory affirms that a receptor is in equilibrium between two states, an active state (on) and an inactive one (off). A ligand may affect this equilibrium by favoring one conformation over the other. For example, an agonist is any ligand that stabilizes the active conformation further.
After detailed studies and accumulating evidences, it is now accepted that this process is more complex. The receptor, as a protein, does not have one or two definite conformations, but it is represented as an energy landscape (93). The activation of any pathway is associated with several conformations that increase the binding of the signaling complexes. After binding a ligand, the receptor traverses to a new energy landscape and different ligands induce different landscapes, thus stabilizing different conformations and exhibiting a preference for different signaling complexes.
Angiotensin type 1A receptor (AT1AR), for example, when stimulated with its
endogenous ligand angiotensin II (Ang II) fails to activate G-protein signaling, while showing a recruitment of β-arrestin2 and activation of ERK1/2 (94).
Another example of the functional selectivity concept is propranolol, which is an inverse agonist on Gαs through the β1-AR, even when it is an agonist activating the MAPK pathway, independent of the G proteins (95).
Previous classifications of ligands as agonists or antagonists have often not taken the potential bias for a particular pathway into account, and assessment of each of these pathways remains to be done. For example, if a ligand can activate the Gα pathway through its receptor,
it is wrong to assume that it can induce the recruitment of β-arrestin as well. Each pathway must be assessed separately to determine the ligand’s effect on each.
With the introduction of functional selectivity, the concept of redundancy has changed. While two distinct ligands can bind the same receptor, they may not activate the same pathways. Although, they might share some physiological functions, they would differ in their signaling capacities.
Biological Implications of Biased Signaling: The importance of biased signaling in the therapeutic field is tremendous. As stated earlier, more than 30% of the prescribed drugs act on GPCRs. It may be beneficial for the treatment of several pathologies, to activate or inhibit only one pathway associated with the targeted receptor and not the others. For example, the µ-opioid receptor is the target of several µ-opioids, such as morphine, which acts as an analgesic through its G-protein activity (96-98). However, activation of β-arrestin by these agonists is associated with respiratory depression. Ideally, we are interested to look at an opioid agonist that is deprived of the ability to recruit the β-arrestin to the receptor.
Another example is the angiotensin receptor and its implication in arterial pressure and cardiovascular diseases. Losartan, an antagonist of this receptor on both Gαq and β-arrestin reduces arterial pressure and cardiac performance. On the other hand, the synthetic biased antagonist TRV120027 on the Gαq activates β-arrestin, leading to several advantages, such as preservation of heart stroke volume and an increase in cardiac performance, giving increased efficiency to the treatment of these patients (99).
Biased Trafficking: Different ligands can lead to a differential phosphorylation by the GRKs (100), which would affect the recruitment of β-arrestin, force, affinity, and duration. As described earlier, β-arrestin plays a role in: (i) the ubiquitination of the receptor, (ii) the assembly of endocytic vesicles, (iii) the trafficking of the receptor and (iv) the initiation of G-independent signaling. Taking that into account, it would be tempting to hypothesize that differential β-arrestin recruitment would lead to biased trafficking of the receptor and subsequent biased signaling through the pathways recruited on the scaffolding protein β-arrestin.
1.12. AMD3100
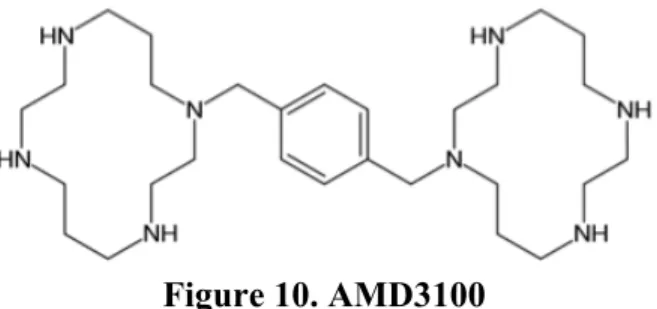
CXCR4 antagonist AMD3100 or Plerixafor is a drug that is used to block the retention of HSCs in the bone marrow and thus mobilize them into the blood stream (101). AMD3100 binds CXCR4 and blocks the effect of the endogenous ligand CXCL12. This molecule is a synthetic one, with a bicyclam structure (Fig. 10) (102). It was first developed to inhibit the HIV 1/2 envelope protein, Gp120, and its cellular entry via the HIV co-receptor (103). During its preclinical evaluation as a potential antiretroviral drug, a significant side effect was observed in otherwise healthy patients (104). Patients receiving intravenous injections of AMD3100 showed abnormally elevated white blood cell counts. As AMD3100 blocks the interaction between CXCR4 and CXCL12, it induces the release of the stem cells in the peripheral blood (105). After the observation of this side effect, research on the medical use of AMD3100 was refocused on another therapeutic indication. Administering AMD3100 in combination with granulocyte-colony stimulating factor (G-CSF) induced rapid mobilization of stem cells into the peripheral circulation (106).
AMD3100 or Plerixafor is currently approved by the FDA for this therapeutic use (72). It is administered to a bone marrow donor, to be able to mobilize HSCs and collect them from the periphery. This is mostly used for autologous stem-cell transplantation in patients with multiple myelomas or non-Hodgkin’s lymphomas (107,108). Prior to the discovery of AMD3100, G-CSF was the standard of care for these patients. For this reason, clinical trials studied the effect of adding AMD3100 to the G-CSF regimen (108).
In addition, Plerixafor is currently used in clinical trials to improve the efficiency of chemotherapy for patients with acute lymphoblastic leukemia (ALL) or acute myeloid leukemia (AML) (109,110). During chemotherapy, cancerous stem cells tend to lodge in the bone marrow environment. This niche, with its stromal cells, provides drug resistance to these cells, which may lead to future relapse (107). Administering Plerixafor is believed to mobilize these cells, and increase their sensitivity to chemotherapy.
Although mobilizing cancer cells from the bone marrow is beneficial in the context of sensitizing them to chemotherapy, inducing the release of cancer cells from their primary
origin increases the risk of metastasis. It has been demonstrated that Plerixafor shows a weak activation on CXCR4. As this receptor is the most widely expressed chemokine receptor on cancer cells (72), this compound activates the migration of these cancer cells. This outcome is devastating as it causes metastatic migration of tumor cells, as well as the contamination of peripheral blood by the cancer cells, leading to subsequent relapse after auto-transplantation with the stem cells (111). For this reason, a thorough understanding of the mechanisms of action of AMD3100 and its effects on CXCR4 signaling is necessary.
Even though AMD3100 is believed to be a neutral antagonist, Zhang et al. have suggested that it can be a partial agonist on the Gαi pathway (85). They have based their deduction after observing that AMD3100 induces GTPγS recruitment to the membrane and a Ca2+ influx, as well as an activation of a galactose gene reporter. Interestingly, these last two pathways are activated by the Gβγ complex rather than the Gαi (112). As discussed earlier, Gαi is the principal α subunit activated by CXCR4, although not the only one. As the complex βγ is shared between different Gα proteins, the activation of any of these can explain the effects observed. For example, activation of Gαq can lead to similar consequences (60). To answer this question, we should assay the signaling effects of Gαi specifically, by observing its effect on the activity of adenylate cyclase (AC).
β-arrestin is a major pathway associated with migration, chemotaxis, and metastasis. Due to the concept of functional selectivity, we cannot deduce the impact of AMD3100 on this pathway based on the previously described experiment. To further understand the role of Plerixafor on cancer metastasis, the effect of this drug on the activation of Gαi and the recruitment of β-arrestin should be studied and is one of the aims of this project.
1.13. TC14012
As mentioned earlier, CXCR4 is an HIV co-receptor. Similar to AMD3100, the peptide T140 was developed specifically as a CXCR4 inhibitor, to exhibit an antiretroviral activity (113). T140 is a peptide of fourteen amino acids, and is derived from self-defense peptides called polyphemusins, which are used by the horseshoe crab (114,115). T140 has shown an anti-HIV activity by antagonizing the cellular entry of the HIV-1 X4-tropic strain (116). T140 showed a high cytotoxicity (115) and was unstable in bovine serum (111). For these two reasons, TC14012 has been developed to increase its pharmacological efficiency (Fig. 11) (117). Two residues of the peptide T140 have been substituted by Citrulline and Deoxy-Citrulline, respectively (111), leading to a decrease in the cytotoxicity of the peptide. Also, the C-terminal of the compound has been amidated to enhance its stability in the serum (115).
Figure 11. TC14012
Analogs of T140 showed several clinical potentials in the treatment of arthritis, cancer, and leukemia (115). One of these analogs, 4F-benzoyl-TN14003 showed anti-metastatic properties by decreasing the migration of breast cancer cells, MDA-MB-231 (118).
The peptide TC14012 was identified to be a CXCR4 inverse agonist on the G-protein pathway (85). This was demonstrated through the same experiments discussed earlier, which implied that AMD3100 be a partial agonist. Nevertheless, inverse agonism effects were not specifically observed on the Gαi pathway, and again its effect on β-arrestin recruitment remains to be elucidated. These two questions are among the aims of this study.
Being an inverse agonist, TC14012 may be more advantageous than AMD3100. As partial agonists like AMD3100 have the capacity to activate and promote the migration of cancer cells expressing the CXCR4 receptor, inverse agonists will demonstrate a clinical advantage by limiting the adverse effects discussed above (115).
2. OBJECTIVES
The main aim of this project is to study the relationship between β-arrestin2 and Gαi on the receptor CXCR4. To achieve this we used previously described mutants affecting the conserved DRY motif and the asparagine 119. Specifically, we had six questions that we wanted to assess in this project:
1. These mutants have only been studied for their capacity to activate the G-proteins in general. Here, we wanted to measure a parameter that is specific to the Gαi activity, such as the concentration of cAMP.
2. Some mutants were previously known to be unable to activate the G-proteins. Whether they were able to activate a G-independent pathway has not been tested. Here, the capacity to recruit β-arrestin2 is assayed and the conformation of this recruitment is compared between the mutants.
3. Signaling and trafficking are interrelated. Keeping in mind that these mutants have a different pattern of signaling, we wanted to assess their respective localization, to find out whether this differential signaling is leading to a differential trafficking.
4. CXCR4 can both activate the Gαi pathway, and recruit β-arrestin2. The concept of functional selectivity states that a ligand can activate one pathway without the other. We wanted to verify if this is true for CXCR4 and determine if β-arrestin2 is dependent on Gαi. This was performed by inhibiting the Gαi pathway with pertussis toxin, while testing for the effect of this inhibition on β-arrestin2 recruitment.
5. To differentiate between an antagonist and an inverse agonist effects, a receptor showing constitutive activity on the pathway is required. N119S has been previously identified as a CAM on Gαi pathway. Yet, no mutant is known to constitutively recruit β-arrestin2 and we aimed to identify such a mutant.
6. The effect of the two synthetic CXCR4 ligands, AMD3100 and TC14012 on CXCR4 signaling has only been studied on the G-proteins pathway. Here we wanted to assay their role on the Gαi signaling specifically, and the recruitment of β-arrestin2, and to differentiate if they were partial agonist, neutral antagonist, or inverse agonist on each of the two pathways.
3. MATERIAL AND METHODS
3.1. Materials and Plasmids
HEK293E cells (Invitrogen, Carlsbad, CA) were cultured in DMEM (4.5 g/L D-Glucose) (Gibco®, Grand Island, NY), FBS (WisEnt, Rocklin, CA), Penicillin/Streptomycin (Invitrogen), G-418 sulfate (WisEnt), and Plasmocin (Invivogen, San Diego, CA). For transfections, OptiMEM (Invitrogen) was used in combination with Polyethylenimine (Polysciences, Warrington, PA). The BRET experiments were conducted in BRET 96-well microplates (PerkinElmer, Waltham, MA) that are coated with 0.1% Poly-D-Lysine (Sigma-Aldrich, St. Louis, MO). BRET tampon contained BSA Fraction V (WisEnt), and the Luciferase substrate Coelenterazine H (Nanolight technology, Pinetop, AZ). Imaging was performed in 8-well chambered coverglass dishes (Labtek, Nunc). Five ligands were used: forskolin (Sigma), Pertussis Toxin (Calbiochem, San Diego, CA), recombined CXCL12 (PeproTech, Rocky Hill, NJ), AMD3100 (Sigma), and synthesized TC14012, a gift from Nobutaka Fujii (111).
Mutants of CXCR4 were developed by the Kunkel method and were previously described (84).
3.2. Cell Culture and Transfection
HEK293E cells were cultured at 37˚C, and 5% CO2 in Dulbecco’s modified Eagle’s
medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 200µg/mL G-418 sulfate, 2.5µg/mL plasmocyin and 100units/mL penicillin/streptomycin. Transfection was conducted on the cells in 6-well plates, 24h after plating 800,000 cells in each well, and a total DNA amount of 2µg was added. The transfection protocol, using polyethyleneimine, has been previously described (119).
3.3. cAMP Biosensor BRET
2Assay
GFP10-Epac-Rluc3 was used as a BRET2 reporter for cAMP reduction (119). Live HEK293E cells were transiently cotransfected with 0.04µg of the Epac plasmid and 1µg of the CXCR4-myc wild-type or mutant. The Epac protein contained both the BRET2 donor and acceptor. 24 hours post-transfection, cells were transferred to white, 96-well, clear-bottom microplates coated with poly-D-Lysine hydrobromide. 36-48 hours post-transfection, the cell medium was changed to BRET buffer (PBS, 0.5mM MgCl2 and 0.01% BSA). Level of
fluorescence was measured by Mithras LB940 instrument (Berthold Technologies) using an excitation filter of 400nm and an emission filter of 515nm. Rluc3 substrate, coelenterazine 400A was added to a concentration of 5µM. Cells were stimulated with 20µM of forskolin, with the different doses of ligand for 10min at room temperature before measurements. Total luminescence was equally taken by Mithras LB940 immediately before the BRET2 measurements. Raw BRET2 was calculated as the ratio of emission of GFP10 at 515nm to the emission of Rluc3 at 400nm (17).
3.4. β-Arrestin2 Recruitment BRET
2Assay
β-arrestin2 recruitment to the CXCR4 receptors was measured using a BRET2 assay
(120). The experiment was conducted similarly to the Epac reporter, while cotransfecting with 0.05µg of each CXCR4 mutant or wild-type tagged with the BRET2 donor Renilla Luciferase 3, and 1µg of β-arrestin2 tagged with GFP10, the BRET2 acceptor. Cells were incubated with the ligands for 5 min at 37˚C and 10 min at room temperature before measurements. Values were corrected by subtracting a background ratio detected when the CXCR4-Rluc3 was transfected alone, giving the BRET2 net values.
3.5. Titration Assays
Similar to the β-arrestin2 recruitment assays, cells were cotransfected with 0.05µg of CXCR4-Rluc3, wild-type, R134A, or N119S with a varying quantity of β-arrestin2-GFP10, between 0.05µg and 1.95µg. 48h post-transfection, cells were stimulated with a saturating
dose of CXCL12, 200nM. Values were corrected by subtracting a background ratio detected when the CXCR4-Rluc3 was transfected alone, giving the BRET2 net values.
3.6. Imaging of CXCR4 Using Spinning Disc Confocal Microscopy
HEK293E cells were transfected with 1µg of CXCR4-YFP, wild-type or mutants, in a pcDNA3 plasmid 48h before imaging. After 24h, cells were transferred to 8-well chambered coverglass dishes. Live cell imaging of transfected cells was performed at 37°C and 5% CO2
using an Ultra view Vox spinning disc confocal system (Perkin Elmer), and an Orca-R2 CCD camera (Hamamatsu). This was combined to a Leica DMI6000B inverted microscope equipped with a motorized piezo-electric stage (Applied Scientific Instrumentation), and using a scanning unit CSU-X1 (Yokogawa) as described earlier (121).
Volocity 5 software (Improvision/PerkinElmer) was used for image acquisition, quantitation, and analysis. Imaging was performed using a Plan Apo 40x (0.85 NA) air objective and a camera binning of 2x2. The YFP tag is observed at 488nm.
3.7. Data Analysis
Collected data were analyzed using Prism 6.0 software (GraphPad Software, San Diego, CA). For the Gαi activation and the β-arrestin2 recruitment, data were fitted with a sigmoidal dose-response curve to determine the potency and the efficiency. Michaelis-Menten curve was used for the titration experiments and the maximum association and the affinity were determined. Differences between the ligands were analyzed using a non-matched one-way ANOVA, followed by a Dunnett’s posttest.
3.8. The Principle of Bioluminescence Resonance Energy Transfer (BRET)
Bioluminescence Resonance Energy Transfer or BRET is a method used to study protein–protein interactions in a live cell medium (122). It can also reflect changes in the intramolecular conformation. This energy transfer is not radioactive, and it occurs between two electromagnetic dipoles, one belonging to the energy donor bioluminescent molecule and the other to the receptor fluorescent molecule (123). However, for this transfer to occur, the two molecules need to be close to each other, with a distance less than 100 Å (124). In addition, some compatibility between the two molecules is required. The emission spectrum of the donor must somewhat overlap the excitation spectrum of the second molecule. Thus, the efficacy of this transfer and intensity of the BRET signal depends on the distance separating the two molecules, as well as the angle between them (122).One widely used bioluminescent donor is the enzyme Renilla luciferase. This enzyme belongs to the animal Renilla reniformis, a sea animal that has a natural capacity for bioluminescence (123). For the experiments described here a variation of this enzyme, Rluc3, is used. This enzyme contains three specific mutations, A55T, C124A, and M185V enhancing its stability and its light output (119,125). This enzyme generates a wavelength of 400 nm by oxidizing its substrate Bisdeoxycoelenterazine (coelenterazine-400a), formally called DeepBlueC. This emission can be transferred to the BRET2 acceptor GFP10, a variation of the
Green Fluorescent Protein. If the two tags are in proximity an efficient transfer of energy can occur and the BRET2 acceptor tag will fluorescence at 515 nm (101).
The BRET technique can demonstrate an intramolecular conformation change, the binding of two proteins or the change of conformation of this interaction (126). The ability to identify intramolecular conformational changes by BRET was used here in the Epac experiment (Fig. 12). The two BRET tags, Rluc3 and GFP10 are both fused to the same protein, Exchange protein directly activated by cAMP (Epac), one on the N-terminal and the other on the C-terminal, respectively. The conformation changes of this protein induce changes in distance and angle between the two tags, which translate into BRET2 value changes (127,128). This protein changes its conformation depending on the cAMP concentration. In its
basal state, the two BRET2 tags are close enough to generate a signal. However, the binding of cAMP induces a conformational change leading to the separation of the BRET2 tags and the
attenuation of the BRET signal.
Furthermore, to study the interaction between two proteins, a BRET signal is perceived when the two proteins, fused with the BRET tags, come into proximity of a distance of less than 100 Å (Fig. 13) (72). Here, the CXCR4 mutants have been fused with Rluc3 and β-arrestin2 with GFP10. In principle, these two proteins do not interact in a cell without a specific stimulation. However, after stimulation with CXCL12, a specific interaction occurs between the two proteins and a BRET signal is generated.
BRET measures resulting from a specific interaction are quantifiable. To achieve this, the ratio of the concentrations of GFP10 over Rluc3 should be controlled. In the case of an unspecific interaction, the signal results from random collisions between the two BRET tags, and an increase in either concentration would increase the chance of such collisions. Based on this, the relation between the BRET signal and the ratio GFP10/RLuc3 follows a straight line (129).
On the other hand, if a specific interaction occurs between the two proteins, BRET signal increases with the GFP10 concentration, when the Rluc3 concentration is held constant (Fig. 14) (111). Still on higher ratios of GFP10/RLuc3, the signal saturates and attains a maximum. This plateau corresponds to a point where all the BRET donor proteins tagged with Rluc3 interact specifically with the BRET acceptor tagged with GFP10. Adding more BRET acceptor proteins will not increase the signal, due to saturation of BRET donors. Thus, this maximal BRET signal, BRETmax, depends on the number of maximum specific interactions
that can be formed.
The BRET50 can be defined as the ratio of GFP10/Rluc3 required to give a BRET signal
that is 50% of BRETmax. This parameter represents the affinity between the two proteins. On
the other hand, BRETmax depends mostly on the distance separating the two BRET tags and