HAL Id: hal-02785621
https://hal.inrae.fr/hal-02785621
Submitted on 4 Jun 2020
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Marin Duthoit
To cite this version:
Marin Duthoit. Génération de remaniements chromosomiques par la méthode CRISPR-Cas9. Autre [q-bio.OT]. 2018. �hal-02785621�
UNIVERSITÉ DE POITIERS
UFR SCIENCES FONDAMENTALES ET APPLIQUÉES
40 Avenue du Recteur Pineau - 80222 POITIERS Cedex Mention
Biologie Santé Science du Médicament
Spécialité
Recherche Ingénierie Biologie Santé
Parcours
GENIE CELLULAIRE
Année 2017-2018
MÉMOIRE DE STAGE de 1
èreannée
présenté par :
Marin Duthoit
TITRE :
Mise au point de la méthode CRISPR Cas9 pour la génération de remaniements chromosomiques in vitro
Stage effectué au Laboratoire de INRA UMR 1388 GenPhySE (23, chemin des Capelles 31076 Toulouse, 05 61 28 50 09)
Sous la Direction de Alain Pinton du 09/04/2018 au 31/07/2018
Remerciements
Je voudrais tout d’abord remercier mon maître de stage Alain PINTON pour son accueil, son suivi et sa confiance. Merci énormément pour la liberté d’action que j’avais, ainsi que pour ses conseils.
Merci à Valérie Fillon, Alain Ducos et Nicolas Mary pour leur accueil au sein de l’équipe et leurs conseils.
Un énorme merci à Nathalie M. pour ses corrections de caryotypes et l’identification d’une délétion.
Merci à Anne et Nathalie P. pour leurs conseils et leur bonne humeur.
Merci à Clémence et Camille pour leur accompagnement, les discussions sur les sujets les plus divers et cette bonne humeur constante.
Encore merci à tous les membres de l’équipe cytogénétique de l’ENVT pour leur accueil chaleureux et l’ambiance familiale du labo qui ont rendu ce stage si agréable.
Enfin, je tiens à remercier toutes les personnes qui m'ont conseillé et qui m’ont aidé lors de la rédaction de ce rapport.
Avant propos
L’Institut National de Recherche Agronomique (INRA) est un organisme de recherche scientifique public sous la double tutelle des ministères en charge de la recherche et de l’agriculture. C’est le premier institut agronomique en Europe et le second au niveau mondial. Au service d’enjeux de société majeurs dans le domaine de l’alimentation, de l’agriculture et de l’environnement, les recherches effectuées permettent de choisir et d’innover à plusieurs niveaux (environnementaux, sanitaires, politiques, territoriaux et sociaux) dans un contexte de compétitivité et de développement durable.
L’UMR (Unité Mixte de Recherche) GenPhySE (Génétique Physiologie et Systèmes d’Elevage) regroupe des personnels ENVT (Ecole Nationale Vétérinaire de Toulouse), ENSAT (Ecole Nationale Supérieure Agronomique de Toulouse) et INRA. Cette unité dépend des départements de Génétique Animale et Physiologie Animale et Systèmes d’Elevage. Les travaux réalisés dans cette UMR se situent dans le domaine de la génétique animale, de la biologie intégrative et des sciences animales. Leurs missions concernent la gestion et l’étude de l’évolution des populations animales d’intérêt agronomique ainsi que l’évaluation et la conception des systèmes d’élevage durables de demain.
L’équipe Cytogène travaille sur 4 axes de recherche : l’architecture du noyau et l’expression du génome, le génome et la plasticité épigénétique pendant la gamétogénèse, la caractérisation de la diversité génétique des populations d’abeilles françaises et le dernier axe sur lequel j’ai pu travailler, la structure génétique et cytogénétique des populations bovines et porcines.
Le laboratoire se trouve sur le site de l’ENVT. Il comprend une plateforme de recherche et une plateforme de contrôle chromosomique des populations animales. La mission de cette dernière est de réaliser un contrôle chromosomique de mammifères afin de détecter la présence d’anomalies chromosomiques. Cela permet notamment d’éviter la diffusion de ces dernières, potentiellement à l’origine de troubles reproductibles dans les élevages porcins et bovins. L’activité de recherche développée au laboratoire consiste à développer des projets qui visent à étudier les effets des anomalies sur le déroulement et les produits de la méiose.
Sommaire
1- Introduction
4
2- Matériel et méthodes
6
a. Structure des plasmides utilisés pour l’édition de génome 6
b.Design et ajout des séquences guides aux plasmides
7
c.
Production, extraction et purification des plasmides
9
d.
Transfection de cellules fœtales de porc
10
e.
Marquage en bandes GTG et caryotypage de mitose
13
3- Résultats
13
a. Production, purification et validation des plasmides
13
b. Transfection et sélection des cellules fœtales
14
c. Caryotypes
15
4- Discussion
17
5- Conclusion
19
6- Bibliographie
20
7- Annexe
22
8- Résumé
39
1- Introduction
Nous savons depuis récemment que l’organisation 3D du génome n’est pas aléatoire et est complexe et dynamique (Bickmore, W. A. & van Steensel, B. 2013 ; Sexton, T. & Cavalli, G. 2015 ; Pombo, A. & Dillon, N. 2015). Cette organisation 3D conditionne les relations de proximité prépondérantes pour diverses fonctions telles que la transcription, la réplication, les réparations de l’ADN ou encore la genèse de translocations chromosomiques (Therizols, P. et al. 2014 ; Gonzalez-Sandoval, A. et al. 2015).
Si l’état de compaction le plus basique, dit nucléosome, est assez bien décrit(Luger, K. et al. 1997), la façon dont il interagit avec d’autres nucléosome reste incertaine. Les interactions dans la chromatine sont essentielles pour l’identité de la cellule. Parmi ces interactions, certaines impliquent notamment des boucles et repliements formés entre des éléments régulateurs, permettant par exemple le rapprochement d’enhancer avec les gènes sur lesquels ils agissent. Mais la façon dont ces interactions sont établies et régulées reste incomprise. Récemment, il a été découvert qu’en plus de ces boucles, la chromatine est organisée en des domaines structurels distincts, qui pourraient représenter des unités fonctionnelles du génome (Sexton, T. T. et al. 2012 ; Nora, E. P. et al. 2012).
Bien que l’architecture tri-dimensionnelle de l’ADN doit être stable, il est aussi nécessaire qu’elle soit assez flexible pour permettre des changements de conformation. De récentes publications suggèrent que, si la structure globale de l’ADN reste robuste à des perturbations lors du développement, des gènes isolés peuvent alterner facilement entre des compartiments actifs et inactifs du génome. De la même façon, les interactions intra et inter domaine de la chromatine peuvent varier fréquemment(Dixon, J. R. et al. 2015). Avec la publication de cartes d’interaction de la chromatine à très haute résolution (Rao, S. S. P. et al. 2014 ; Schuettengruber, B. et al. 2014), il est apparut que l’organisation de la chromatine est plus complexe qu’on ne le pensait précédemment. Ainsi, l’organisation des sous-domaines ou les interactions entre les promoteurs ont été révélées à l’aide de séquençage profond ou avec de nouvelles techniques.
En effet, l’analyse de l’organisation de la chromatine peut être analysée en combinant différentes approches basées sur la microscopie (limitée aux régions d’intérêt) et sur la capture de la conformation chromosomique (3C, HiC) (analyse tout génome) (Bonev, B. & Cavalli,G. 2016).
Les analyses de type HiC ont permis de mettre en évidence l’existence de domaines particuliers du génome ; les TAD (topologically associating domains) (Rao, S. S. P. et al. 2014 ; Dixon, J. R. et al. 2012 ; Nora, E. P. et al. Heard, E. 2013).
Ces domaines rassemblent la majeure partie des interactions entre enhancer et promoteurs.
structurels dans les génomes (structural variations ou SV) et quantitatifs, incluant les délétions, duplications, inversions, insertions et translocations (Weischenfeldt, J. et al. 2013). Ces réarrangements sont susceptibles d’altérer l’organisation de la chromatine et par conséquent les interactions et donc le fonctionnement des génomes.
Ainsi, un résumé de la littérature (Spielmann, M et al. 2018) suppose que lorsque les SV ont lieu en dehors de ces TAD, l’impact sur l’expression des gènes serait nul, ou du moins mineur. Un SV dans un TAD provoquerait une variation de l’expression des gènes concernés, alors qu’un SV ayant lieu à la jonction entre deux TAD mènerait à l’apparition de nouvelles interactions enhancer-promoteurs, en fonction des cas on parlera alors de « fused TAD », « Neo-TAD » ou « Shuffled TAD » (Annexe 1).
Afin de mieux comprendre les relations entre structure et fonctionnement du génome, le laboratoire d’accueil développe un nouveau projet de recherche sur l’impact des réarrangements chromosomiques sur l’organisation et l’activité du génome.
Dans le cadre de ce projet, le travail réalisé visait à développer une méthode permettant de remanier à façon le génome en générant des réarrangements chromosomiques (translocations,…)
Afin d’obtenir des translocations chromosomique, des cellules fœtales de porc ont été éditées via Crispr-Cas9. La Cas9 génère des cassures double-brin dans des zones précises du génome définies à l’avance, permettant alors une recombinaison des segments chromosomiques et la génération de réarrangements.
Pour cela, des plasmides codant pour la Cas9 ont été produits. Différents ARN guides ont été clonés dans ces plasmides en fonction des zones visées.
Ces plasmides ont ensuite été transfectés seuls ou par paires dans des cellules fœtales de porc. Après une phase de sélection et de culture, les cellules ont été prélevées en phase de mitose pour observer la présence ou non de réarrangements via la réalisation de caryotypes.
Dans la littérature (Torres, R. et al. 2014 ; Lagutina, I.V. et al. 2015 ; Jiang, J. et al. 2016 ; Blasco, R.B. et al. 2014), la technique a fonctionné sur des cellules humaines et murines, avec une réussite d’environ 1%, nous chercherons à savoir si la technique fonctionne également sur le porc, et si oui, avec quelle spécificité.
2- Matériel et méthodes
a. Structure des plasmides utilisés pour l’édition de génome
Les plasmides sont de petits fragments d’ADN (moins de 10kb) circulaires, présents généralement dans les bactéries.
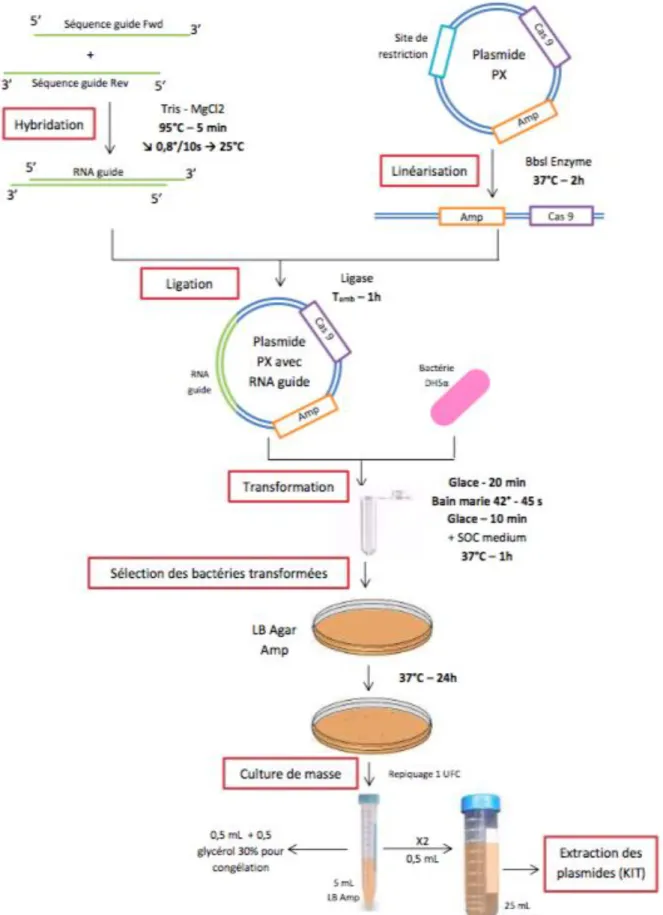
Les plasmides utilisés pour les transformations bactériennes sont construits à partir des plasmides PX458 et PX459 de chez Addgene (Figure 1) (Ran,F.A. et al. 2013). Chacun d’eux est composé de :
- Une séquence codant la Cas9, enzyme (endonucléase) coupant à la base près dans un endroit précis et défini par avance du génome.
- Un site de restriction BbsI au niveau du promoteur hU6 permettant d’insérer les séquences ARN guides préalablement choisies.
- Un site ORI étant l’origine de réplication, une séquence unique permettant l’initiation de la réplication du plasmide.
- Deux moyens de sélection ;
o Une séquence codant une résistance à l’ampicilline (antibiotique) permettant la sélection des bactéries recombinées après transformation avec le plasmide. o Pour le plasmide PX458 : une séquence codant pour l’EGFP (Enhanced Green Fluorescent Protein), une protéine fluorescente verte issue d’une méduse (Aequorea victoria). Après transfection, les cellules ayant intégrées le plasmide deviennent fluorescentes sous lampe à vapeur de mercure.
o Pour le plasmide PX459 : une séquence codant une résistance à la Puromycine. Les cellules ayant intégrées le plasmide peuvent être sélectionnées par ajout de puromycine dans le milieu de culture. Cette dernière est un antibiotique inhibiteur de la synthèse des protéines produit par Streptomyces alboniger. Elle remplace la partie 3’ d’un aminoacyl-ARNt, ce qui provoque l’arrêt anticipé de la traduction par le ribosome.
b. Design et ajout des séquences guides aux plasmides
La séquence du génome complet du porc est aujourd’hui entièrement connue et disponible librement sur internet. Le design des séquences guides se fait à partir de ces données. Dans un premier temps, la séquence d’ADN contenant le point de cassure voulu est recherchée dans ces banques de données. Puis, grâce au logiciel en ligne CRISPOR (http://crispor.tefor.net/), en choisissant le type d’animal (Sus Scrofa) et le type de nucléase utilisé (20 Bp-NGG-Spcas9), plusieurs séquences possibles de guide de 20 paires de bases sont proposées. Cette séquence doit être unique dans le génome du porc et donc spécifique afin de limiter les Off-target (coupure sur des zones non voulues de l’ADN). Elle doit également précéder directement une séquence PAM (Protospacer Adjacent Motif) spécifique à l’endonucléase utilisée (5’ NGG 3’ pour la Cas9 de Streptococcus pyogenes) et permettant à la cas9 de couper l’ADN.
La spécificité des ARNguides a été testée en faisant un BLAST de leur séquence sur le génome du porc (Annexes 24 à 36)
Une fois designées, les séquences guides doivent être synthétisées puis insérées dans les plasmides au niveau du site de restriction BbsI (Figure 2).
Dans un premier temps, il faut hybrider les séquences guides reverse et forward. Cette hybridation permet la création de bouts cohésifs permettant une meilleure insertion des guides dans le plasmide. En parallèle, la linéarisation du plasmide grâce à des enzymes de restriction au niveau du site de restriction BbsI permet d’ouvrir le plasmide pour l’insertion des séquences guides.
Il faut ensuite mettre en présence les plasmides ouverts, les séquences guides hybridées et une enzyme de ligation, la ligase, pour recirculariser le plasmide contenant la séquence guide.
Ces étapes de préparation des plasmides auront été réalisées en amont du stage.
La nomenclature des guides utilisés lors de ce stage permet d’identifier le chromosome et la région visée. Il y a en général deux guides différents par région, et chacun a été combiné avec le plasmide 458 (8) et le plasmide 459 (9) ; numéro du chromosome-région-version du guide (1 ou 2)- type de plasmide (8 ou 9)
c. Production, extraction et purification des plasmides
L’utilisation d’une bactérie, E.Coli, permet d’obtenir un grand nombre de plasmides. Pour chaque guide, une transformation par choc thermique (20 min sur glace, 45s au bain marie à 42°C puis 10 min sur glace, avant de laisser reposer 1h avec du milieu SOC) est alors réalisée avant d’étaler les bactéries sur gélose avec ampicilline. Cette étape permet de sélectionner uniquement les clones ayant intégré le plasmide. Après 24h de culture, des clones sont récupérés à l’aide d’une anse puis mis en préculture dans des tubes 4ml de LB Amp. Après une demi journée de préculture, les 4ml sont divisés dans 4 tubes de 25ml de LB AMP.
Une extraction et une purification des plasmides sont ensuite réalisées gra ce a un Kit d’extraction NucléoBond (Macherey Nagel).
Les plasmides sont ensuite purifiés à l’aide d’une colonne fonctionnant sur le principe d’échanges d’ions. L’ADN (ici les plasmides) chargé négativement se fixe de manière hautement spécifique sur les groupements MEA (méthyl-amino-éthanol) chargés positivement de la colonne.
L’élution permet de séparer les oligonucléotides de longueurs différentes, des protéines, carbohydrates et autres déchets cellulaires. Le pH alcalin permet d’éluer les protéines, les polysaccharides, les métabolites et les tri-nucléotides. Puis des concentrations croissantes en sel permettent de séparer l’ARN et l’ADN simples et doubles brins des plasmides circulaires.
Enfin, un séquençage des plasmides obtenus est réalisé pour confirmer la présence de la séquence guide.
Ce protocole a été réalisé pour la production de 25 plasmides et guides différents, coupant sur les chromosomes 1, 13, 14 et 15 (Annexes 22 à 36). Pour la suite des expérimentations, des plasmides produits au préalable au laboratoire seront également à disposition (Annexes 22 et 23).
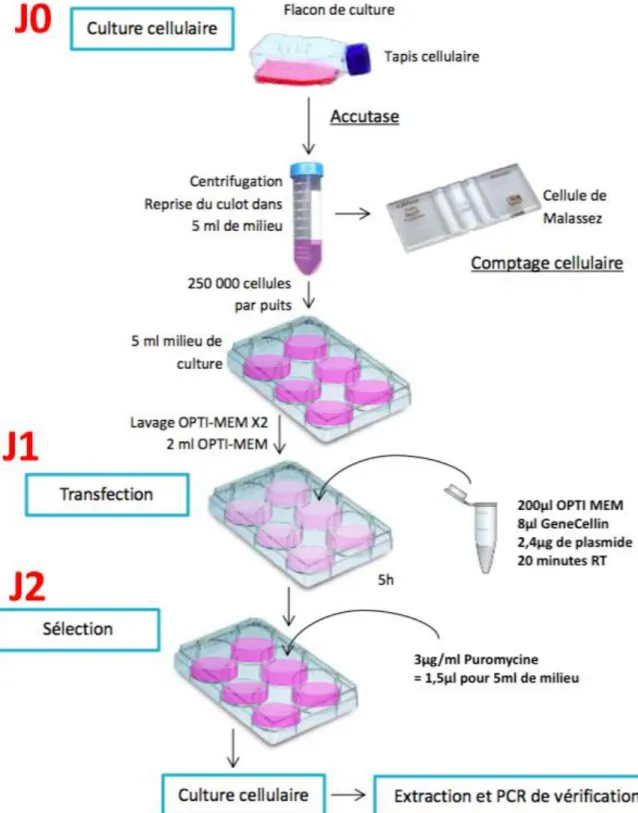
d. Transfection de cellules fœtales de porc
Les cellules utilisées sont cultivées à 37°C avec 5% de CO2. Le milieu utilisé est du DMEM-F12 additionné de 20% de SVF (Sérum de veau Fœtal), de 1% d’une solution anti-bactérienne/anti-mycotique, de 1% de pyruvate de sodium et de 1% d’acides aminés non essentiels.
Afin de réaliser la transfection (Figure 3), les cellules sont ensemencées, après décollement à l’accutase (15min de traitement à 37°C), dans une boite 6 puits à 200 000 - 250 000 cellules par puits.
Le lendemain, les puits sont lavés deux fois à l’OPTI-MEM. Les puits sont remplis avec 2ml d’OPTI-MEM avant de recevoir la solution de transfection, à savoir :
- 200µl OPTI-MEM - 8µl GeneCellin
- 2,4µg de plasmide (si 2 guides utilisés, 1,2µg de chaque)
Cette solution aura été préparée et laissée incuber au moins 20min avant le traitement. Le Genecellin est un composé de polymères dits nanoparticules. Ces polymères vont encapsider l’ADN et ce complexe ADN/polymères pénètre dans la cellule par un mécanisme d’endocytose. Contrairement à des agents lipidiques tels que la lipofectamine, le Genecellin ne désorganise pas les lipides de la membrane plasmique, ce qui le rend théoriquement moins toxique pour la cellule.
Au bout de 5h, les puits sont lavés deux fois avec du milieu de culture, avant de reprendre les cellules dans 5ml de milieu.
Le 3e jour, les cellules ayant intégré un plasmide GFP (PX458) fluorescent en vert sous une lampe à vapeur de mercure. Dans le cas de l’utilisation d’un plasmide codant pour la résistance à la puromycine (PX459), une étape de sélection sur deux cycles de 24h est ajoutée (1,5µl Puromycine pour 5ml de milieu, soit 3µg/ml).
A l’issu de cette étape en puromycine, les cellules présentes sont censées avoir intégré soit le PX459, soit le PX459 et le PX458 dans le cas d’une double transfection.
Les cellules sont cultivées dans les puits jusqu’à confluence puis passées successivement en flasque de culture à 25cm2, 70cm2 puis 2 boites de 70cm2.
La méthode a été réalisée pour des transfections simples ainsi que pour des doubles transfections (2 guides différents). Dans le cas de transfections simples, la méthode génère une cassure seulement sur un chromosome. On devrait alors observer des chromosomes avec une délétion, ainsi que des chromosomes ayant obtenu le fragment perdu. Dans le cas d’une double transfection, une translocation réciproque a plus de chance de se produire. Dans le cas particulier de l’utilisation du guide 14-68 (ARN guide non spécifique choisi dans une région répétée), on peut s’attendre à l’apparition de nombreux fragments, ou à des chromosomes très endommagés (Annexe 30).
Les transfections réalisées sont récapitulées dans le tableau 1
Afin de vérifier la présence de translocations induites, un caryotypage systématique des mitoses est réalisé. Cette méthode permet notamment de vérifier visuellement la présence de translocations en classant les chromosomes en fonction de leur taille, de la position du centromère et des bandes observées après une coloration au Giemsa.
Pour ce marquage, les cellules sont récupérées en étape de mitose, soit en tapant les flacons de cultures et en récupérant le milieu de culture (les cellules en mitoses sont moins solidaires au support de culture), soit en ajoutant de la colchicine dans le milieu ; cette dernière méthode permet de bloquer les cellules en métaphase en perturbant le fuseau de division.
Les cellules obtenues sont ensuite soumises à un choc hypotonique avec du sérum de veau nouveau né au 1/6e, ce choc permet de dilater les noyaux des cellules. Elles sont ensuite préfixées et fixées avec du fixateur de Carnoy (3 volumes d’éthanol 100% pour 1 volume d’acide acétique 100%).
Les cellules sont ensuite étalées sur lame, puis marquées en bandes GTG (dénaturation à la trypsine et coloration au giemsa). Après coloration, les zones riches en Adénine et en Thymine apparaissent foncées au microscope à contraste de phase, tandis que les zones riches en Guanine et Cytosine restent claires.
Les lames sont ensuite scannées au GSL120 (automate permettant de sélectionner et capturer des structures sur une lame, l’élément recherché est paramétré via une base d’image) puis traitées pour obtenir le caryotype via le logiciel CytoVision (Leica).
3- Résultats
a. Production, purification et validation des plasmides
Lors de la culture des bactéries transformées à l’aide des plasmides PX458 et PX459, il a été constaté que le nombre de clones recombinants étaient beaucoup plus élevé avec le plasmide PX459 (environ 60 clones par boite de Petri) qu’avec le plasmide PX458 (entre 0 et 5 clones par boite). Pour certains plasmides PX458, une seconde transformation a été nécessaire pour obtenir des clones et passer à l’étape suivante, l’un d’entre eux n’a pas pris du tout.
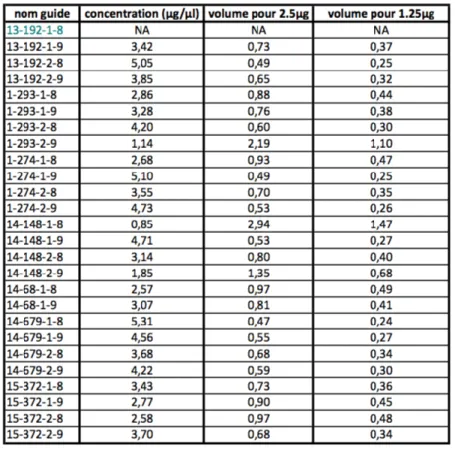
Lors des purifications, des valeurs finales de concentration très variées ont été obtenues ; allant de 0,85µg/µl à 5,31µg/µl (Tableau 2). Les solutions présentant des impuretés n’ont pas été utilisées pour la suite des expérimentations.
Le séquençage se fait par envoi à GATC Biotech de 5µl de plasmide à 80-100ng/µl additionnés de 5µl d’amorces à 5µM. Tous les plasmides utilisés pour la suite des expériences ont été validés comme contenant la bonne séquence guide.
Tableau 2 : Concentration des guides purifiés et volumes utilisés pour les transfections
b. Transfection et sélection des cellules fœtales
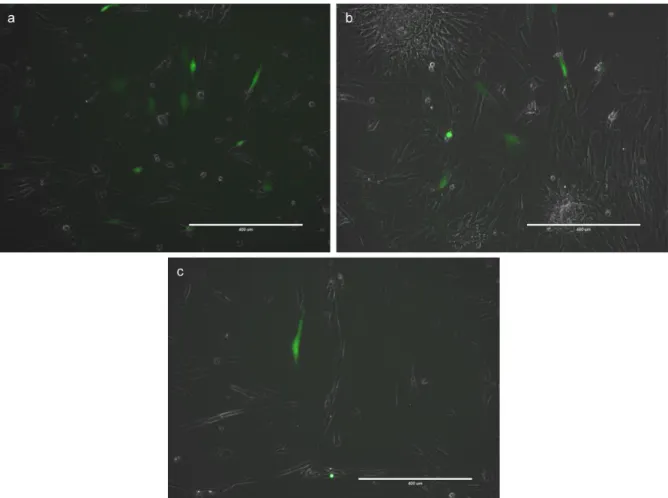
Afin d’estimer l’efficacité des transfections, les cellules sont observées au microscope x10 au contraste de phase (repérage des cellules) puis avec un filtre GFP afin de déterminer le taux de cellules GFP+, ayant intégré le plasmide PX458 (Figure 4).
Bien que le nombre de transfections réalisées n’était pas assez élevé pour établir un lien clair, il semble que l’efficacité de transfection dépende du guide utilisé. En effet, certaines constructions induisent plus de cellules GFP+ que d’autres. Ainsi, les plasmides contenant les guides 14-68-1-8 et 1-274-1-8 semblent être plus efficaces que d’autres comme celui contenant le guide 14-148-1-8 (Tableau 3). Ce dernier a notamment été utilisé sur 8 transfections, dont 2 seulement ont présenté des signaux GFP.
En ce qui concerne la transfection avec le plasmide PX459, il est apparut que des cellules normales survivaient à la sélection à la puromycine dans les puits témoins. Ce nombre variait en fonction des amas cellulaires présents dans les puits. En effet, les cellules fœtales semblent se regrouper par amas lors des ensemencements. Cette conformation semble protéger les cellules du traitement à la puromycine.
c. Caryotypes
Au total, sur l’ensemble des transfections, le caryotypage de 436 cellules sélectionnées de façon aléatoire a permis d’identifier 49 anomalies, de 19 types différents (Annexe 2 à 21 et Tableau 4).
La prévalence de certaines de ces anomalies est relativement élevée en comparaison avec les données de la littérature. En effet, alors que cette technique est décrite avec une réussite d’environ 1% dans les cellules humaines ou murines, la translocation 10-1 (guide 14-148-1-9) et l’anomalie du chromosome 3 (guides 15-372-2-8/13-192-1-14-148-1-9) ont été observées ici avec des fréquences respectives de 15,6% et 9,2%.
Les cellules issues des transfections n’utilisant que le guide 14-68-1-8 n’ont pas été caryotypées pour cause de mauvaise qualité des métaphases obtenues. Si le caryotypage était impossible, toutes les images obtenues (environ 180) ont cependant été comptées afin de déceler des anomalies. En effet, ce guide a été édité spécialement pour viser une centaine de points dans le génome (Annexe 30), les images ont donc été analysées dans le but d’identifier la présence de fragments ou de délétions nombreuses sur les chromosomes 14 et 7 (facilement identifiables) de ces cellules. Lors de ces comptages, aucune anomalie n’a été relevée.
Figure 5 : Translocation entre les chromosomes 10-1 via le guide 14-148-1-9
4- Discussion
Lors des transformations bactériennes avec le plasmide PX458, il est apparu que le nombre de clones qui se développaient était très faible en comparaison de celles avec PX459. Il est possible que pour des raisons inconnues, PX458 soit difficile à intégrer pour les cellules. Si les plasmides purifiés ont systématiquement été séquencés avant le début du stage et possèdent tous la séquence guide insérée, il est possible que le plasmide 458 utilisé ne soit pas de bonne qualité ou ait été lésé d’une façon ou d’une autre, influant sur l’efficacité de la ligation. Cela pourrait expliquer les variations de transformation entre 458 et 459, ainsi que les variations dans l’expression de la GFP via les différents guides 458. De même, cela permettrait de comprendre pourquoi 14-148-1-9 induit de nombreux réarrangements alors que 14-148-1-8 ne produit pas de GFP (pas de caryotypes disponibles pour le moment pour ce guide).
Des caryotypes de cellules transfectées par 14-148-1-8 permettront de savoir si le défaut du plasmide 458, joue sur l’efficacité de transfection des cellules, ou seulement sur l’expression de la GFP.
Comme dit précédemment, un autre élément est apparu lors de la première transfection, des cellules témoins ont survécu au traitement à la puromycine. Une gamme de puromycine de 1µl à 3,5µl par puits (de 2µg/ml à 7µg/ml) a permis d’établir que quelle que soit la concentration utilisée, des cellules survivaient, et que ce nombre variait peu. Les lames étudiées par la suite présenteront donc un certain nombre de cellules normales.
Lors de l’étape de caryotypage, nous nous sommes également aperçu que la qualité des lames empêchait d’avoir un nombre de cellules « caryotypables » élevé. Il a été supposé que le bain de trypsine de la coloration était trop long. Cette partie du protocole sera modifiée pour de prochaines analyses. Le nombre de cellules utilisables, ainsi que la qualité des caryotypes utilisés est à prendre en compte dans l’analyse ; il est possible que certaines anomalies soient plus fréquentes qu’il ne semble. Par exemple, la translocation entre les chromosomes 10 et 1 n’apparaît qu’une 1 fois pour 13 caryotypes réalisés lors de la transfection 15-372-1-8/1-274-2-9.
La qualité des lames a également impacté le temps nécessaire au caryotypage des images et leur analyse. Une grande majorité des cellules étaient ainsi difficilement exploitables, présentant des chromosomes sans bandes ou très flous (Annexe 37). En temps normal, un caryotype est réalisé en 5 à 8 minutes par les techniciennes expérimentées de la plateforme d’analyse. Les caryotypes utilisés lors pour ce rapport ont tous été réalisés individuellement et manuellement par l’expérimentateur puis validés par ces techniciennes. Ils représentent chacun environ 20minutes. Cela correspond à environ 145 heures, soit un quart de la période de stage. Améliorer la méthode au niveau des bains de trypsine permettra, en plus d’obtenir de meilleures images et une meilleure analyse, de gagner du temps sur le caryotypage.
En dehors de la qualité des images obtenues, plusieurs profils de caryotype ont été obtenus ; les images sans anomalies, celles avec une délétion sur un chromosome et celles présentant une translocation.
Il peut y avoir 2 raisons à une absence d’anomalie ; soit la cellule analysée correspond à une cellule où la Cas9 n’a pas coupé l’ADN (valable également dans le cas d’une cellule normale ayant survécu à la puromycine), soit la Cas9 a coupé mais il n’y a pas eu de réarrangement. Des tests menés préalablement au laboratoire ont montré en utilisant la T7 qu’il pouvait y avoir cassure dans les régions ciblées sans générer de translocation, avec cependant des réarrangements dans d’autres régions suggérant la formation de cassures doubles brins multiples (dans et hors la région ciblée). La formation de cassures multiples hors cibles est connue et des méthodes pour identifier ces off-target sont décrites dans la littérature (exemple : Tsai, S. Q. et al. 2015). Cette méthode a notamment permis de décrire jusqu’à 150 off-target sur des expériences de CRISPR-Cas9.
Cela pourrait également expliquer certaines anomalies et translocations obtenues. Ainsi, les séquences des guides 15-372-2-8 et 13-192-1-9 ne devaient pas toucher le chromosome 3 (Annexes 15, 28 et 36), anomalie qui est cependant la plus présente avec cette transfection. Même raisonnement avec la translocation 10-1 (Annexes 2 et 31).
Selon les données de la littérature, la genèse des réarrangements chromosomiques est influencée par différents paramètres : position des chromosomes dans le noyau (organisation 3D du génome), affinité moléculaire entre les différentes régions, état de compaction, disponibilité de la chromatine. Il semblerait même qu’en fonction de l’endroit où a lieu la cassure ou la réparation dans le noyau, les mécanismes de réparation sont plus ou moins efficaces (Bonev, B. & Cavalli,G. 2016 ; Spielmann, M et al. 2018 ; McCord, R.P & Balajee, A. 2018 ; Boroviak, K. et al. 2017).
On peut donc émettre l’hypothèse que dans le cas de cassures double brin multiples, ce sont les paramètres précédemment décrits qui influenceront l’apparition de tel ou tel type de réarrangements.
En ce qui concerne les translocations obtenues, certaines semblent revenir avec des transfections qui n’ont aucun guide en commun ; notamment les translocations 10-1 et 13-1. La première apparaît avec les transfections 14-148-1-8 et 15-372-1-8/1-274-2-9, tandis que la seconde apparaît lors des transfections 14-148-1-8 et 15-372-2-8/13-192-1-8. Une analyse plus précise qu’un caryotype, comme un séquençage, permettrait d’identifier la zone à l’origine du remaniement, permettant ainsi de déterminer si ces translocations sont identiques. Si oui, elles pourraient ne pas avoir été causée par les transfections. Pour le moment, étant donné la fréquence d’apparition de la translocation 10-1 parmi les cellules transfectées avec le guide 14-148-1-8, cette dernière est supposée spécifique de ce guide.
5- Conclusion
Nos résultats montrent que le système CRISPR-Cas9 est efficace et répétable chez le porc pour générer des remaniements chromosomiques. Toutefois, l’apparition de réarrangements en dehors des régions ciblées suggère, en cohérence avec certaines données de la littérature, que la spécificité de la technique CRISPR-Cas9 n’est pas toujours optimale.
Dans notre travail, nous avons fait le choix de caryotyper systématiquement toutes les cellules analysables. Ce travail, relativement lourd et consommateur de temps, a cependant permis de détecter des anomalies chromosomiques en dehors des régions ciblées montrant le manque de spécificité de la méthode. Une analyse PCR (amplification des régions transloquées) classiquement utilisée pour la détection des réarrangements après édition via CRISPR-Cas9 n’aurait pas permis de détecter ces remaniements hors cibles. On peut donc légitimement se poser la question si la recherche par PCR de réarrangements induits par CRISPR-Cas9 est vraiment optimale pour vérifier la spécificité de la méthode et ne présente pas un risque de ne pas détecter certaines anomalies hors cibles.
Afin de produire des clones et des remaniements de façon plus efficace, il est envisagé de travailler par la suite sur des IPS et d’ensemencer les cellules après transfection dans des plaques 96 puits (1 à 2 cellules par puits). Il serait alors possible d’isoler et de générer des lignées de cellule remaniées et d’étudier les effets de ces divers remaniements sur les fonctions du génome.
Il serait également intéressant d’utiliser d’autres nucléases compatible avec le système Crispr, telle que la Cas12 (ou Cpf1). Si son mécanisme diffère et reste peu connu, cette enzyme semble très prometteuse (Jeon, Y. et al. 2018).
Un projet supplémentaire du laboratoire suite à la génération de ces lignées remaniées, serait de tenter de réparer de façon spécifique les anomalies produites précédemment, de façon similaire à ce qui a déjà été décrit dans la littérature (Lekomtsev, S. et al. 2016).
6- Bibliographie
1- Bickmore, W. A. & van Steensel, B. (2013). Genome architecture: domain organization of interphase chromosomes. Cell 152, 1270–1284.
2- Sexton, T. & Cavalli, G. (2015).The role of chromosome domains in shaping the functional genome. Cell 160, 1049–1059.
3- Pombo, A. & Dillon, N. (2015). Three-dimensional genome architecture: players and mechanisms. Nat. Rev. Mol. Cell Biol 16, 245–257.
4- Therizols, P. et al. (2014). Chromatin decondensation is sufficient to alter nuclear organization in embryonic stem cells. Science 346, 1238–1242.
5- Gonzalez-Sandoval, A. et al. (2015). Perinuclear anchoring of H3K9 - Methylated chromatin stabilizes induced cell fate in C. elegans embryos. Cell 163, 1333–1347. 6- Luger, K. et al. (1997). Crystal structure of the nucleosome core particle at 2.8 Å
resolution. Nature 389, 251–260.
7- Sexton, T. T. et al. (2012). Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 148, 458–472.
8- Nora, E. P. et al. (2012). Spatial partitioning of the regulatory landscape of the X – inactivation centre. Nature 485, 381–385.
9- Dixon, J. R. et al. (2015). Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336.
10- Weischenfeldt, J. et al. (2013). Phenotypic impact of genomic structural variation: insights from and for human disease. Nature Reviews Genetics 14, 125–138.
11- Rao, S. S. P. et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680.
12- Schuettengruber, B. et al. (2014). Cooperativity, specificity, and evolutionary stability of Polycomb targeting in Drosophila. Cell Rep 9, 219–233.
13- Bonev, B. & Cavalli,G. (2016). Organization and function of the 3D genome. Nature
Reviews Genetics, 17, 661-677.
14- Dixon, J. R. et al. (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380.
15- Nora, E. P., Dekker, J. & Heard, E. (2013). Segmental folding of chromosomes: a basis for structural and regulatory chromosomal neighborhoods? Bioessays 35, 818–828. 16- Spielmann, M et al. (2018). Structural variation in the 3D genome, Nature Reviews
Genetics.
17- Torres, R. et al. (2014). Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nature communications
4964, 1-8.
18- Lagutina, I.V. et al. (2015). Modeling of the Human Alveolar Rhabdomyosarcoma Pax3-Foxo1 Chromosome Translocation in Mouse Myoblasts Using CRISPR- Cas9 Nuclease. PLoS Genet 11(2), 1-24.
19- Jiang, J. et al. (2016). Induction of site-specific chromosomal translocations in embryonic stem cells by CRISPR/Cas9. Scientific Reports 6, 1-9.
20- Blasco, R.B. et al. (2014). Simple and Rapid In Vivo Generation of Chromosomal Rearrangements using CRISPR/Cas9 Technology. Cell Reports 9, 1219–1227.
Chromosome Translocation Pattern. Chromosome Translocation, Advances in
Experimental Medicine and Biology 1044, 113-133.
22- Boroviak, K. et al. (2017). Revealing hidden complexities of genomic réarrangement generated with Cas9. Scientific Reports 7, 1-8.
23- Tsai, S.Q. et al. (2015). GUIDE-Seq enables génome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33, 187-197.
24- Lekomtsev, S. et al. (2016). Efficient generation and reversion of chromosomal translocations using CRISPR/Cas technology. BMC Genomics, 1-6.
25- Ran,F.A. et al. (2013). Genome engineering using the CRISPR-Cas9 system. Nature
Protocols 8, 2281-2308.
26- Jeon, Y. et al (2018). Direct observation of DNA target searching and cleavage by CRISPR-Cas12a. Nature Communications 9.
7- Annexe
Annexe 2 : Translocation entre les chromosomes 10-1 via le guide 14-148-1-9
Annexe 4 : Translocation entre les chromosomes 13-1 via le guide 14-148-1-9
Annexe 6 : Anomalie du haut du chromosome 14 via les guides 14-679-2-8/1-274-2-9
Annexe 8 : Anomalie du chromosome 1 via les guides 14-679-2-8/1-274-2-9
Annexe 10 : Anomalie du chromosome 9 via les guides 14-679-2-8/1-274-2-9
Annexe 12 : Anomalie du chromosome 13 via les guides 14-679-2-8/1-274-2-9
Annexe 14 : Translocation entre les chromosomes 10-1 via les guides 15-372-1-8/1-274-2-9
Annexe 16 : Translocation des chromosome 13-1 via les guides 15-372-2-8/13-192-1-9
Annexe 18 : Translocation des chromosomes 13-14 via les guides 15-372-2-8/13-192-1-9
Annexe 20 : Anomalie du chromosome 14 via les guides 15-372-2-8/13-192-1-9
Annexe 24 : Carte de spécificité du guide 1-274-1
Annexe 23 : Tableau récapitulatif guides et séquences stage 2017
Annexe 22 : Tableau récapitulatif guides et séquences stage 2018
Annexe 25 : Carte de spécificité du guide 1-274-2
Annexe 26 : Carte de spécificité du guide 1-293-1
Annexe 27 : Carte de spécificité du guide 1-293-2
Annexe 29 : Carte de spécificité du guide 13-192-2
Annexe 30 : Carte de spécificité du guide 14-68-1 (100 hits)
Annexe 31 : Carte de spécificité du guide 14-148-1
Annexe 33 : Carte de spécificité du guide 14-679-1
Annexe 34 : Carte de spécificité du guide 14-679-2
Annexe 35 : Carte de spécificité du guide 15-372-1
Annexe 37 : Comparaison de caryotypes "de mauvaise qualité" (a et b) en comparaison avec un caryotype "de qualité correcte" (c)
Annexe 38 : Réactifs utilisés, composition et rôles Annexe 39 : Confection d’une source de sourires bourrative
C’est en cette suite d’étapes que le fondant au chocolat tu confectionneras Pour cette recette les éléments suivants rassembler tu devras ;
2 hectogrammes de beurre, de sucre et de chocolat noir à pâtisser 1 hectogramme 25 de moulure de blé, 4 œufs à casser.
Dans une marmite, à feu doux le beurre et le chocolat tu liquéfieras
Dans un bol à part, les œufs jaunes et blancs confondus, tu mélangeras au sucre et au blé. Quand dans le faitout liquide ce sera, les deux préparations dans le bol tu devras
|| mixtionnioner. Dans un plat la pâte tu étaleras, à 200°C tu enfourneras,
Pendant 15 minutes attendre tu devras,
A partir de là, toutes les 5 minutes la cuisson tu vérifieras.
Le sortir de l’étuve tu devras, en fonction de la texture qui te sierras. Il ne me reste qu’à te souhaiter,
Il est connu aujourd’hui que l’ADN présente différents niveaux de compaction permettant diverses fonctions telles que la transcription, la réplication, les réparations de l’ADN ou encore la genèse de translocations chromosomiques. Ces variations de compaction jouent également un rôle sur les différentes interactions existant entre les gènes, leurs promoteurs et des régions non-codantes. Des domaines particuliers où se rassemblent la majeure partie des interactions entre enhancer et promoteurs ont ainsi été définis. Il a été observé dans la littérature que lorsque des réarrangements structurels avaient lieu dans ces domaines, de nouvelles interactions apparaissaient. Afin de mieux comprendre les relations entre structure et fonctionnement du génome, un nouveau projet de recherche a été développé. Ce dernier porte sur l’impact des réarrangements chromosomiques sur l’organisation et l’activité du génome. L’objectif du travail réalisé était de mettre au point une méthode permettant de remanier à façon le génome en générant des réarrangements chromosomiques. Pour cela, des cellules fœtales de porc ont été éditées via Crispr-Cas9 avec différentes séquences guides. Les cellules ainsi éditées ont ensuite été caryotypées après coloration au Giemsa. Suite à ces expérimentations, 19 anomalies différentes ont été obtenues. Parmi elles, certaines avaient une fréquence très élevée (15,6% ou 9,2% par exemple au lieu de 1% dans la littérature).
Ces résultats tendent à montrer l’efficacité de la technique pour éditer des cellules de porc. It is known that DNA has different levels of compaction allowing various functions such as transcription, replication, DNA repair or the genesis of chromosomal translocations. These variations of compaction also have a rôle in the different interactions between genes, theirs promoters and non-coding regions. Specific domains where most of these interactions are concentrated have been defined. Some studies have already shown that when structural réarrangements take place in these domains, new interactions appeared. In order to better understand the links between structure and working of the genome, a new project has been developed. It focuses on the impact of chromosomal réarrangement on the organization and activity of the genome. The goal of this study is to develop a method for designing the genome by generating chromosomal rearrangements. To do so, porcine fetal cells were edited with Crispr-Cas9 with different guide sequences. Then, the edited cells were karyotyped after Giemsa staining. 19 different anomalies have been obtained by these experiments. Among them, some had a very high frequency ; 15.6% or 9.2% for example instead of 1% in the literature.
These results tends to show the efficiency of the technique for editing pig cells.