HAL Id: tel-01686126
https://tel.archives-ouvertes.fr/tel-01686126
Submitted on 17 Jan 2018
HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Emma Goux
To cite this version:
Emma Goux. Utilisation d’aptamères impliqués dans des complexe “ kissing ” pour la détection de petits ligands. Sciences pharmaceutiques. Université Grenoble Alpes, 2016. Français. �NNT : 2016GREAVO46�. �tel-01686126�
THÈSE
Pour obtenir le grade de
DOCTEUR DE LA COMMUNAUTÉ UNIVERSITÉ
GRENOBLE ALPES
Spécialité : Sciences du Médicament
Arrêté ministériel : 7 août 2006
Présentée par
Emma GOUX
Thèse dirigée par Eric PEYRIN Co-dirigée par Corinne RAVELET et Co-encadrée par Valérie GUIEU
préparée au sein du Département de Pharmacochimie Moléculaire dans l'École Doctorale Chimie et Sciences du Vivant
Utilisation d’aptamères impliqués dans
des complexes « kissing » pour la
détection de petits ligands
Thèse soutenue publiquement le « 27 octobre 2016 », devant le jury composé de :
Pr Eric DEFRANCQ
Professeur, Université Grenoble Alpes (Président du jury)
Pr Claire DEMESMAY-GUILHIN
Professeur, Université de Lyon 1 (Rapporteur)
Dr Frédéric DUCONGE
Chercheur, CEA (Rapporteur)
Dr Benoît LIMOGES
Directeur de recherche, Université Paris Diderot (Examinateur)
Dr Jean-Jacques TOULME
Directeur de recherche émérite, Université de Bordeaux (Examinateur)
Pr Eric PEYRIN
Professeur, Université Grenoble Alpes (Directeur)
Dr Corinne RAVELET
Maitre de conférence, Université Grenoble Alpes (Co-directrice)
Dr Valérie GUIEU
2 Je tiens ici à transmettre mes plus sincères remerciements à mes très chers directeurs et encadrants Pr Eric Peyrin, Dr Corinne Ravelet et Dr Valérie Guieu pour leur présence, leurs conseils et leur réactivité. Je tiens également à remercier chaleureusement les membres du jury le Pr Demesmay, Dr Ducongé, le Pr Defrancq, Dr Limoges et Dr Toulmé pour avoir accepté d’évaluer ce travail.
Je voudrais également remercier toute l’équipe NOVA pour leur aide et leurs conseils en commençant par Emmanuelle Fiore et Dr Sandrine Perrier. A tout le personnel du DPM avec qui j’ai vécu pendant 4 ans des moments inoubliables : les dégustations de vins les concerts et les soirées endiablées avec le Girls Power (Florine, Flavia, Noémie, Roseline et Dounia), les bières et les « Je vais mourir ! » avec Vinisius, les enguelades avec Samuele, les cours d’arabe et de chimie organique avec Ikram et Amel, les randonnées pendant lesquelles ma vitesse décroit exponentiellement, les repas de Noël, chandeleur, pâques … les weekends en Italie et en Belgique avec Florine, Dounia, Flavia, Cédric, Samuele et Laurent. Sans oublier Denis, Annabelle, Luc, Antoine, Farid, Delphine, Véronique, Pascale, Angèle, Antoine, Catherine, Florence, Cécile, Ben, Romain, Jean-Robert, Jeff, Julien, Kim, Luma, Momo, Simon, et tous les autres. A cette ville de Grenoble où l’on meurt de froid l’hiver et crève de chaud l’été mais où l’on mange si bien ! Je terminerai en remerciant ma famille, en particulier ma Mère qui a corrigé mes fautes d’orthographe, mon homme Bernard qui me soutient et me rend la vie si douce et tous mes amis pour leur amour. Je souhaite donc dédier ce travail à toutes ces personnes sans qui je ne serais pas celle que je suis aujourd’hui.
Abréviations 3
Abréviations
Abréviation Signification A Adénine AC Anticorps ADN Acide désoxyribonucléique ADO Adénosine ADOsw Aptaswitch anti-adénosine AMP Adénosine monophosphateANSES Agence nationale de sécurité sanitaire de l’alimentation, de l’environnement et du travail
apt Aptamère ARN Acide ribonucléique ARNm ARN messager ARNt ARN de transfert ATP Adénosine triphosphate AuNP Nanoparticule d'or BA Agent bloquant BC Brin complémentaire BNA Acide nucléique ponté bp Paire de bases BSPP Bis(p-sulfonatophényl)phénylphosphine C Cytosine CE Electrophorèse capillaire CEC Electrochromatographie capillaire CTP Cytidine triphosphate D Dispersion DDII Deep dark quencher II DIS Site de dimérisation DMLA Dégénérescence maculaire liée à l'âge ELAKCA Enzyme linked aptamer kissing complex assay ELISA Enzyme linked immunosorbent assay F Fluorescéine FA Anisotropie de fluorescence
4 FDA Food and Drug Administration FP Polarisation de fluorescence G Guanine GTP Guanosine triphosphate GTPsw Aptaswitch anti-guanosine triphosphate Homo-RET Transfert d'énergie par résonnance entre deux molécules identiques HRP Péroxydase de raifort HyCbl Hydroxycobalamine IgE Immunoglobuline E IUPAC Union internationale de chimie pure et appliquée KD Constante de dissociation LC Chromatographie en phase liquide LNA Acide nucléique verrouillé LOD Limite de détection LSP Plasmon de surface localisé MRE Elément de reconnaissance moléculaire NC Nucléocapside NP Nanoparticule NP-apt Nanoparticule fonctionnalisée avec des aptamères NP-BC Nanoparticule fonctionnalisée avec des brins complémentaires NP-kiss Nanoparticule fonctionnalisée avec des aptakisses NP-sw Nanoparticule fonctionnalisée avec des aptaswitches nt Nucléotide PCR Réaction de polymérase en chaine PDGF Facteur de croissance de plaquette-dérivé PEG Polyéthylène glycol PMT Tube photomultiplicateur PNA Acide nucléique peptidique PVDF Fluorure de polyvinyllidène QD Quantum dot QCM Microbalance à cristaux de quartz RET Transfert d'énergie par résonance RMN Résonance magnétique nucléaire SELEX Systematic Evolution of Ligand by EXponential enrichment
Abréviations 5 SEO Oligonucléotide amplificateur de signal SP Plasmon de surface SPR Résonance des plasmons de surface T Thymine TAR Région de réponse trans-activante TCEP Tris(2-carboxyéthyl)phosphine TEM Microscopie électronique à transmission THE Théophylline THEsw Aptaswitch anti-théophylline Tm Température de fusion TR Texas Red U Uracile UNA Acide nucléique déverrouillé UTP Uridine triphosphate UTR Région non transcrite UV Ultra-violet VEGF Facteur de croissance de l'endothélium vasculaire VIH Virus d'immunodéficience humain IR Infra rouge
6
Liste des figures
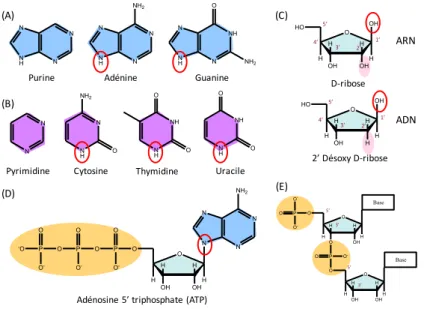
n° de figure Titre et légendes p. Figure 2-1: structures chimiques des bases puriques (A) et pyrimidiques (B), des riboses et désoxyriboses (C), d’un exemple de nucléotide – l’adénosine triphosphate – (D) et d’une chaine d’ARN (E) (en bleu et en violet respectivement noyaux puriques et pyrimidiques, en rose C2’, en vert ribose et en orange groupement phosphate, entouré en rouge emplacement de la liaison N-glycosidique).
18
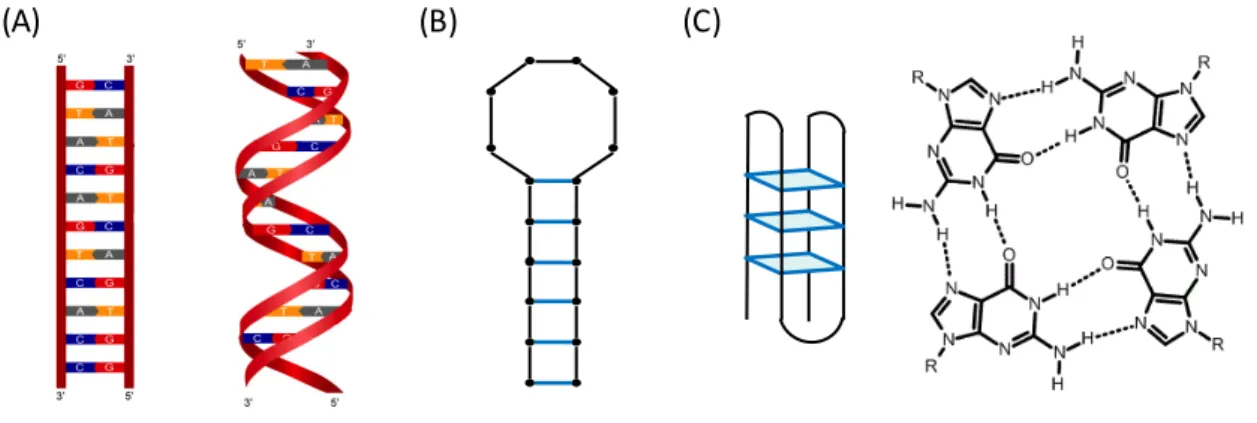
Figure 2-2 : (A) structure secondaire et modélisation 3D d’un duplex d’ADN, (B) structure d’une hairpin et (C) exemple de structure d’un brin formant des G-quadruplex, et schéma des liaisons hydrogène formées entre les 4 guanines du plan. 19 Figure 2-3 : construction des séquences de la banque aléatoire pour la sélection des aptamères 21 Figure 2-4 : schéma du protocole SELEX 21 Figure 2-5 : (A) électrophérogrammes de l’analyse d’échantillon de sérum reconstitué dopé en IgE (concentration en IgE : 0-100-400 nM de droite à gauche ; A* aptamère), (B) électrophérogramme de l’analyse d’un mélange de L- et D-arginine et (C) chromatogramme de la purification de la L-sélectine (flèche changement de la composition de la phase mobile pour l’élution de la protéine cible). Adaptée des références 68–70,72
24
Figure 2-6 : exemples de modifications chimiques de l’ADN et de l’ARN pour améliorer leur stabilité. Adaptée des références 20,21 26
Figure 2-7 : comparaison des structures du D-ARN et du L-ARN. 26
Figure 2-8
: (A) structure secondaire de l’aptamère anti-adénosine en présence (en bas) et en absence (en haut) de cibles (AI et AII sont les cibles dans le site I et le site II respectivement ; paire de bases de type Watson-Crick (○) et paire de bases non canonique(●)), (B) structure 3D de l’aptamère anti-adénosine en présence d’AMP (en rouge atome de phosphore, en blanc autres atomes de l’aptamère et en jaune autres atomes de l’AMP) et (C) représentation en ruban de la structure 3D de l’aptamère anti-adénosine en présence de l’AMP – le squelette est montré en ruban et les bases et paires de bases sont symbolisées en cylindres (en violet la poche de fixation des AMP et les deux tiges sont mises en bleu et vert, les AMP complexés sont en blanc sauf les atomes de phosphore qui sont marqués en rouge) adapté de la référence 86. 28
Listes des figures et tableaux 7 Figure 2-9 : (A) structure secondaire de l’aptamère anti-théophylline en absence de cible (paire de bases de type Watson-Crick (○) et paire de bases non canonique(●)), (B) structure secondaire de l’aptamère anti-théophylline en présence de cible (lignes violette, rose, bleu et orange triplets, théophylline (T), ligne rouge squelette ribose phosphodiester, paire de bases de type Watson-Crick (○) et paire de bases non canonique(●)), (C) représentation bâton de la structure 3D de l’aptamère anti-théophylline en présence de cible (en violet les tiges haute et basse, en bleu et vert poche de fixation de la théophylline, en vert entouré en noir la théophylline, ligne rouge squelette ribose phosphodiester) adapté de la référence 99
30
Figure 2-10
: (A) templates ADN des deux types de séquences utilisés pour la sélection de l’aptamère anti-GTP (N : nucléotide), (B) structure secondaire de l’aptamère anti-GTP en absence de cible (paire de bases de type Watson-Crick (○) et paire de bases non canonique(●)), (C) structure secondaire de l’aptamère anti-GTP en présence de cible (lignes bleue et orange triplets, GTP vert GTP cible, ligne rouge squelette ribose phosphodiester, paire de bases de type Watson-Crick (○) et paire de bases non canonique(●)), (C) représentation bâton de la structure 3D de l’aptamère anti-GTP en présence de cible (en vert et orange les tiges haute et basse respectivement, en bleu et jaune poche de fixation du GTP, entouré en noir le GTP cible) adapté des références 104,106
32
Figure 2-11 : distribution des états excités pour une population de fluorophores immobiles adaptée de la référence 108 35
Figure 2-12
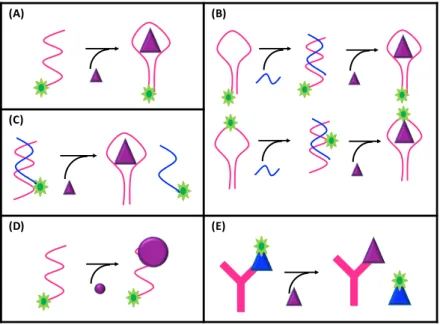
Exemples de plateformes de détection par anisotropie de fluorescence. Ligne rose l’aptamère, ligne bleue brin complémentaire, anticorps (Y), cible macromoléculaire (●), petit ligand ( ), analogue structural ( ), fluorophores ( ). (A) essai non compétitif pour petite molécule1,2, (B) essai non compétitif pour petite molécule assisté par SEO3, (C) essai compétitif pour petite molécule4,5, (D) essai non compétitif pour macromolécule6–8, (E) immunoessai compétitif pour petite molécule. Adapté des références 9–11
38
Figure 2-13 : spectres d’absorption des solutions colloïdales de nanoparticules d’or à différents stades de coalescence adapté de la référence 113 40 Figure 2-14 : séquences de l’hairpin DIS du VIH-1 adapté de la référence 130 44 Figure 2-15 : mécanisme de formation du VIH-1 adapté de la référence 138 45 Figure 2-16 : (A) structure secondaire du riboswitch adénine et (B) et (C) modélisation 3D de la conformation fermée du riboswitch en présence de cible adapté de la référence 136. Les couleurs sont les mêmes que sur le schéma (A) et en surface pointillée les ions Mg2+ hydratés 47
Figure 2-17 : (A) structure secondaire du riboswitch HyCbl et (B) structures 3D des conformations ouverte et fermée en absence et en présence de cible respectivement adapté de la référence 143. La boucle L5 en rouge et le site de fixation du ribosome (RBS) en vert 47 Figure 2-18 : formation du kissing complex entre les séquences CopA et CopT adapté de la référence 130 48
8 Figure 2-19 : structure secondaire (A) avant et (B) après formation des kissing complexes adapté de la référence 130. En rouge les bases complémentaires impliquées dans les kissing complexes, sous-lignés en vert le site de fixation du ribosome et le codon AUG et encadré en bleu le site de fixation de la protéine Hfq 48
Figure 2-20 : structure des kissing complexes formés entre (A) l’aptamère anti-TAR et l’hairpin TAR du VIH-1 adapté de la référence 145 et (B) les aptamères anti-ARN du virus de l’hépatite C et les hairpins SL1 et Domaine IV adapté de la référence 146
49
Figure 2-21
: principe du schéma d’analyse basé sur l’utilisation d’aptamère impliqué dans un kissing complex pour la détection de petites molécules. (a) introduction de la séquence d’une boucle (en rouge sur le schéma) capable de former un kissing complex avec l’aptakiss (en violet) à la place de la boucle initiale de l’aptamère.
49
Figure 2-22 : sensorgrammes SPR des systèmes anti-GTP et anti-adénosine (A) analyse du GTP à différentes concentrations (de 0 à 1 mM) et (B) analyse de l’adénosine à différentes
concentrations (de 0 à 8 mM) 51
Figure 2-23
: sensorgrammes SPR des systèmes anti-GTP et anti-adénosine : (A) GTPsw1’ en présence d’ATP, (B) ADOsw1’ en présence de l’inosine, (C) et (D) respectivement injection du GTPsw1’ et de l’ADOsw1’ en présence de leur cible sur une puce modifiée K1mut et (E) injection de l’ADOsw1’mut en présence d’adénosine sur une puce modifiée aptakiss.
52
Figure 2-24
: (A) sensorgramme SPR du système anti-adénosine avec les séquences ADOsw1’ rallongées pour une concentration d’adénosine de 8 mM et (B) sensorgramme SPR de l’analyse de l’adénosine à différentes concentrations (de 0 à 8 mM) en présence de l’ADOsw1’-TA GC.
52
Figure 3-1 : spectres d’émission et d’excitation normalisés de la fluorescéine (noir et vert respectivement) et du texas red (bleu et magenta respectivement) 55
Figure 3-2 : principe de l’analyse en multiplex basée sur un schéma sandwich et avec une détection en anisotropie de fluorescence (noté r) avec r1 la valeur de FA du système
1 et r2 la valeur de FA du système 2
56
Figure 3-3 : schéma de la stratégie d’optimisation du schéma d’analyse à double reconnaissance pour la détection de petites molécules. 79 Figure 3-4 : principe de l’analyse colorimétrique basée sur le système sandwich et l’utilisation de nanoparticules d’or 95 Figure 3-5 : principe de l’analyse colorimétrique basée sur un schéma aptamère / BC et l’utilisation de nanoparticules d’or 95
Figure 3-6 : (A) valeurs de r0-rf des systèmes de boucle sw1’/kiss1 et sw3’/kiss3 en absence de cible (n = 3) et (B) valeurs de ∆r pour différents ratio kiss : sw et différentes concentrations d’ADO (gris : 31,25 µM ; bleu : 62,5 µM et blanc : 500 µM ; n = 3)
97
Figure 3-7 : structure chimique du (A) tris(2-carboxyéthyl)phosphine (TCEP) et du (B) Bis(p
Listes des figures et tableaux 9 Figure 3-8 : (A) spectres d’absorption UV-visible normalisés des AuNP greffées aptamère (tirets rouges) et BC (points rouges), du mélange des deux types de particules (violet) et (B) les valeurs de R pour les NP-apt et NP-BC séparés, les NP-aptamère et NP-BC ensemble (n=2)
98
Figure 3-9 : schéma du protocole de greffage d’oligonucléotides thiolés sur nanoparticules d’or 99 Figure 3-10 : les valeurs de ∆R pour les NP-apt et NP-BC séparés, et le mélange les NP-apt/NP-BC en présence ou en absence d’adénosine (n=3) 100
Figure 3-11
: (A) spectres d’absorption UV-visible normalisés des AuNP greffées ADOsw3’-26 (tirets rouges) et K3 (points rouges), du mélange des deux types de particules dans un ratio kiss : sw 1:1 en absence (vert) et en présence de 500 µM d’adénosine (violet), (B) les couleurs correspondantes des solution colloïdale et (C) les valeurs de ∆R pour différents ratio kiss : sw en absence (gris) et en présence (bleu) de cible (500 µM) 101 Figure 3-12 : (A) courbe de calibration de l’adénosine avec le système sandwich (n = 6 ; en encart la partie linéaire de la courbe avec la droite de régression) et (B) étude de la sélectivité du test colorimétrique en présence d’analogue structuraux de l’adénosine à 500 µM (∆R relatif = ∆R / ∆R500 avec ∆R500 la valeur de ∆R pour 500 µM d’adénosine ; n = 3 ; A :adénosine ; I : inosine ; G : guanosine ; X : xanthine ; T : théophylline) 102 Figure 5-1 : schéma du dispositif optique du lecteur microplaque adapté de la référence 167. 110 Figure 5-2 : schéma du système optique du spectrofluorimètre adapté de la référence 168. 1 : lampe au xénon / 1a : bloc d’alimentation de la lampe / 2 : monochromateur d’excitation / 3 : compartiment de l’échantillon / 4 : monochromateur d’émission / 5 : détecteur de signal / 6 : détecteur de référence. 113 Figure 5-3 : schéma du système optique du spectromètre UV-visible à double faisceau adapté de la référence 169 114 Figure 5-4 : étude de la réactivité croisée. A : THEsw4’ + K4-3’TR / B : ADOsw1’ + K1-3’F / C : THEsw4’ + K1-3’F + ADO (250 µM) + THE (250 µM) / D : ADOsw1’ + K4’-3’TR + ADO (250 µM) + THE (250 µM) 117
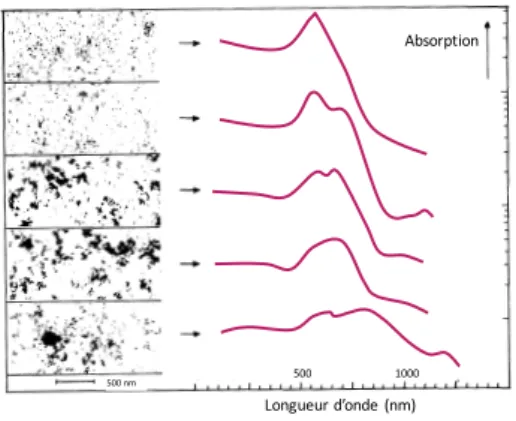
Figure 5-5 : (A) image de nanoparticules d’or obtenu par TEM (échelle : 30 nm) (B) comptage des nanoparticules d’or en fonctions de leur taille (n = 1228) (en rouge distribution
gaussienne) 119
Figure 5-6 : protocole de greffage d’oligonucléotides thiolés sur nanoparticules d’or optimisé adapté de 59 120
Figure 5-7 : test sur le système anti-théophylline (ratio volumique pour le mélange NP-sw/NP-kiss : 1/1 ; ratio volumique pour le mélange + BATHE : 0,78/1 ; concentration en BATHE
10 Figure 5-8 : analyse de l’ADO dans le sérum : comparaison de différents types de traitement (ADO dans l’eau (gris), ADO dans le sérum filtré sur PVDF dilué à 1/20 et 1/50 (rouge et bleu respectivement), ADO dans sérum traité par l’éthanol dilué à 1/20 (vert) et ADO dans sérum filtré sur NanoSep dilué à 1/20 (blanc) 122
Liste des tableaux
n° du tableau Titre et légendes p. Tableau 2-1: exemple de schémas d’analyse basés sur l’utilisation d’aptamère. Ligne rose l’aptamère, ligne bleue brin complémentaire, cible macromoléculaire (
●
), petit ligand ( ), nanoparticules d’or dispersées (●
) et agrégées (●
), graphène oxyde (hexagone noir), puce de mesure (rectangle bleu), fluorophores ( ), fluorophores éteints ( ), quencher (Q).23
Tableau 2-2 : valeur de !0 en fonction de " adapté de la référence 108 35 Tableau 2-3 : séquences des hairpins des couples 1, 2 et 4 (en rouge : boucle). 50 Tableau 2-4 : séquences des aptamères et des aptaswitches (souligné : boucle initiale de l’aptamère ; en bleu : bases supprimées ; en rouge : boucle K1’). 51 Tableau 2-5 : séquences des ADOsw (en rouge : boucle K1’ et en surligné jaune : bases ajoutées dans la tige) 52
Tableau 3-1 : niveaux des facteurs étudiées par plan d’expériences. 100
11
Table des matières
REMERCIEMENTS ... 2ABREVIATIONS ... 3
LISTE DES FIGURES ... 6
LISTE DES TABLEAUX ... 10
TABLE DES MATIERES ... 11
1
INTRODUCTION ... 14
2
REVUE BIBLIOGRAPHIQUE ... 18
2.1
LES APTAMERES : DES OUTILS DE RECONNAISSANCE MOLECULAIRE PROMETTEURS ... 18
2.1.1
Rappels sur l’ADN et l’ARN ... 18
2.1.2
Définitions générales des aptamères ... 20
2.1.2.1
Quel type de cible ? ... 20
2.1.2.2
Mode de sélection des aptamères : le SELEX ... 20
2.1.2.3
Quels usages ? ... 22
2.1.2.4
Stabilisation des acides nucléiques ... 25
2.1.3
Les aptamères modèles ... 27
2.1.3.1
L’aptamère anti-adénosine ... 27
2.1.3.2
L’aptamère anti-théophylline ... 29
2.1.3.3
L’aptamère anti-guanosine triphosphate ... 32
2.2
METHODES DE DETECTION ... 33
2.2.1
L’anisotropie de fluorescence ... 33
2.2.1.1
Rappels sur la théorie de l’anisotropie de fluorescence ... 34
2.2.1.2
Exemples de plateformes de détection ... 36
2.2.2
Utilisation des nanoparticules d’or en colorimétrie ... 39
2.2.2.1
Principe du changement de couleur des nanoparticules d’or ... 39
2.2.2.2
Synthèse et modification des nanoparticules d’or ... 40
2.2.2.3
Techniques de caractérisation ... 41
2.2.2.4
Exemples de plateformes de détection ... 42
2.3
LE « KISSING COMPLEX » : DE SES IMPLICATIONS BIOLOGIQUES A LA DETECTION DE PETITS LIGANDS ... 43
2.3.1
Caractéristiques des kissing complexes ... 43
2.3.2
Rôles biologiques ... 45
2.3.2.1
Dimérisation du génome viral : Cas du VIH-1 ... 45
2.3.2.2
Régulation de l’expression génétique ... 46
2.3.3
Utilisation des kissing complexes pour la détection de petites molécules ... 49
12 2.3.3.1
Schéma d’analyse à double reconnaissance ... 49
2.3.3.2
Sélection des boucles ... 50
2.3.3.3
Design des aptaswitches et détection de l’adénosine et du GTP ... 50
2.3.3.4
Conclusion ... 53
2.4
OBJECTIFS DE LA THESE ... 53
3
RESULTATS ET DISCUSSION ... 55
3.1
MISE EN PLACE DE LA PLATEFORME DE DETECTION EN MULTIPLEX ... 55
3.1.1
Stratégie de développement ... 55
3.1.2
Article n°1 ... 57
3.1.3
Conclusions ... 78
3.2
OPTIMISATION DU SYSTEME DE DETECTION DOUBLE RECONNAISSANCE DE L’ADENOSINE ... 78
3.2.1
Optimisation de l’ADOsw et nouveau schéma d’analyse ... 78
3.2.2
Article n° 2 ... 80
3.2.3
Conclusions ... 94
3.3
DEVELOPPEMENT D’UN TEST COLORIMETRIQUE POUR LA DETECTION DE PETITES MOLECULES ... 94
3.3.1
Principe de l’analyse ... 94
3.3.2
Nouveau système de boucle ... 96
3.3.3
Optimisation du greffage des oligonucléotides sur AuNP ... 97
3.3.3.1
Tests préliminaires ... 97
3.3.3.2
Optimisation des conditions de greffage et d’analyse ... 99
3.3.4
Application du schéma sandwich pour la détection colorimétrique de l’adénosine ... 101
3.3.5
Conclusions ... 102
4
CONCLUSION ET PERSPECTIVES ... 105
5
ANNEXES EXPERIMENTALES ... 109
6
BIBLIOGRAPHIE ... 123
13
14
1 Introduction
Pour aider à la protection de l’environnement et au diagnostic de maladies mais aussi dans les domaines de l’agroalimentaire et de la médecine légale, il est nécessaire de disposer de méthodes d’analyse performantes. Ces méthodes doivent être rapides et peu couteuses afin d’analyser un grand nombre d’échantillons pour détecter la présence éventuelle de la molécule recherchée. Ainsi plusieurs plateformes de bio-analyse ont été développées pour la détection de molécules cibles dans les fluides biologiques, les matrices environnementales ou alimentaires. Elles sont basées sur l’utilisation d’éléments de reconnaissance moléculaire (MRE pour « molecular recognition element »).12 Les MRE les plus utilisés sont les anticorps (AC) car ils ont la capacité de reconnaître leur cible avec une forte affinité et une grande spécificité. Ils sont utilisés pour la détection d’analytes depuis les années 50 et leur utilisation a explosé dans les années 70. Cependant, les inconvénients des AC sont multiples et sont dus à leur méthode d’identification et de production. En effet, les méthodes conventionnelles sont réalisées in vivo chez des animaux ce qui peut poser des difficultés pour l’identification d’AC pour des cibles non immunogéniques et/ou très toxiques. De plus, ces AC ne sont pas facilement utilisables pour des applications thérapeutiques, car ils peuvent provoquer une réaction immunitaire chez l’homme. Par ailleurs, la présence d’AC naturels dans les fluides biologiques peut poser des problèmes lors de diagnostics – ils peuvent se complexer avec les AC MRE de la plateforme et perturber l’analyse. Enfin, l’identification et la production des AC sont longues, fastidieuses, couteuses et soumises aux fluctuations du vivant.13 Du fait de ces problèmes, de plus en plus de chercheurs se tournent vers d’autres MRE tels que les aptamères. Ces derniers ont été développés dans les années 90 par deux équipes de chercheurs américains, Gold dans le Colorado et Szostak dans le Massachusetts.14,15 Ce sont des oligonucléotides simple-brins d’ADN ou d’ARN qui possèdent une grande affinité et une grande spécificité pour une cible donnée.16 On les a surnommés les « anticorps chimiques » car à l’instar des AC naturels ils peuvent être utilisés comme MRE dans un grand nombre de méthodes de diagnostic, d’imagerie et de séparation. Ils sont sélectionnés et synthétisés in vitro contrairement aux AC, ce qui permet la sélection d’aptamère pour une grande variété de cibles allant de l’ion inorganique à la cellule13 et notamment pour des cibles non immunogéniques ou très toxiques. Leur nature synthétique facilite également leur marquage par différents groupements chimiques (fluorophores, motif cholestréryle, bras espaceur …).16 Elle permet également une grande flexibilité dans l’ingénierie post sélection des séquences afin de mieux s’adapter au design des biocapteurs et bio-essais (les aptamères peuvent être tronqués à leur extrémité, scindés en deux …).2,17–19 Il est aussi possible de modifier la nature chimique du squelette de l’aptamère afin d’améliorer sa stabilité dans les milieux complexes tels que les fluides biologiques.20,21 Enfin, les aptamères sont non immunogéniques. Ils peuvent donc être utilisés plus facilement en thérapeutique.13
Chapitre 1 : INTRODUCTION GÉNÉRALE 15 Pour développer des méthodes d’analyses plus spécifiques, des systèmes dits sandwich ont été mis au point utilisant des AC comme MRE. Dans ce type de schémas d’analyse, la cible, une macromolécule ou une entité plus grosse (i.e. une bactérie, un virus ou une spore), est reconnue simultanément par deux AC. Ces schémas sont de type hétérogène : au moins un des AC est immobilisé sur un support solide ou une nanoparticule. Dans le 1er cas, un seul AC est immobilisé sur le support de mesure. Il est appelé AC primaire et sert à capturer la cible et à éliminer la matrice de l’échantillon. Le second AC, dit secondaire, est libre en solution et se fixe sur un autre site de reconnaissance de la cible. Il peut être marqué ou non par un transducteur du signal. Ce type de schémas d’analyse peut être couplé à plusieurs méthodes de détection – méthodes électrochimiques, optiques (e.g. fluorescence) ou sensibles aux variations de masse (microbalance à cristaux de quartz, résonnance des plasmons de surface).22 L’AC secondaire peut notamment être marqué par une enzyme. On parle alors de méthode ELISA (« Enzymatic Linked immunoSorbent Assay »). Par exemple, l’AC secondaire peut être couplé à la péroxydase de raifort (HRP) dont le substrat change de couleur après la réaction enzymatique. Ainsi, plus la concentration en cible est élevée, et plus la couleur de la solution sera intense. Zeng et al. ont, quant à eux, décrit en 2012 un immunoessai dans un format sandwich pour la détection d’antigènes viraux de l’hépatite B dans lequel les AC sont immobilisés sur des nanoparticules.23 L’AC primaire est immobilisé sur un nanobâtonnet d’or et l’AC secondaire est greffé sur un quantum dot (QD) fluorescent. La formation du double complexe immun provoque le rapprochement du QD et du nanobâtonnet d’or ce qui induit l’extinction de la fluorescence. Grâce à ces schémas de type sandwich, il est également possible de mettre en œuvre une méthode d’amplification du signal qui peut, par exemple, être basée sur l’utilisation de nanoparticules22 ou couplée à une amplification par une réaction de polymérase en chaine (PCR) dans le cas où au moins un des MRE utilisés est un oligonucléotide ou est couplé avec un oligonucléotide.24
Cependant, ce type de test nécessite la présence sur la cible de deux sites épitopes bien distincts pour la reconnaissance simultanée de deux MRE, ce qui n’est pas le cas des petits analytes – molécules ayant un poids moléculaire inférieur à 3000 Da. Or, leur détection dans des matrices complexes présente de nombreux intérêts car ces petites molécules peuvent être des biomarqueurs, des drogues, des principes actifs de médicaments ou encore des polluants. C’est dans ce cadre qu’un bio-essai basé sur un schéma sandwich dédié aux petites molécules a été mis au point au laboratoire.18 Il repose sur l’utilisation d’aptamères modifiés pouvant former des « kissing complexes » avec d’autres oligonucléotides. Les « kissing complexes » sont des architectures d’ADN ou d’ARN particulière. Il s’agit de l’interaction de deux séquences au niveau de boucles apicales avec la formation de paires de bases. Les objectifs de la thèse ont été d’étudier et d’optimiser ce nouveau schéma d’analyse, puis de montrer sa potentialité et sa versatilité en le couplant à différents modes de transduction du signal et en mettant en œuvre l’analyse simultanée de plusieurs molécules. Ainsi, mon manuscrit de thèse est divisé en 4 chapitres.
16
La présente introduction constitue le chapitre 1. Le chapitre 2 correspond à une revue bibliographique sur les aptamères en général (leurs cibles, leur méthode de sélection, leurs utilisations), et les aptamères modèles utilisés au cours de ces travaux, sur les méthodes de détection employées (anisotropie de fluorescence et colorimétrie basée sur l’utilisation de nanoparticules d’or) et sur les kissing complexes (caractéristiques et rôles biologiques). La fin de ce chapitre décrit le schéma sandwich pour petites molécules développé par le laboratoire et l’équipe de Jean-Jacques Toulmé du laboratoire ARNA (Université de Bordeaux). Le chapitre 3 est dédié à la partie expérimentale. Il sera subdivisé en 3 sous-chapitres :
Ø le premier détaille la mise en place de l’analyse simultanée de l’adénosine, de la théophylline et de la guanosine triphosphate basée sur le schéma sandwich pour petites molécules.
Ø le second aborde l’optimisation du schéma d’analyse sur le modèle du système de reconnaissance anti-adénosine.
Ø le dernier est consacré au développement d’un test colorimétrique basé sur l’utilisation de nanoparticules d’or modifiée par des oligonucléotides.
Enfin, une conclusion générale et les perspectives du projet (chapitre 4) viendront clore cet exposé. Les données expérimentales sont détaillées en annexe à la fin de ce manuscrit.
17
18
2 Revue bibliographique
2.1 Les aptamères : des outils de reconnaissance moléculaire prometteurs
2.1.1 Rappels sur l’ADN et l’ARNL’ADN – acide désoxyribonucléique – et l’ARN – acide ribonucléique – sont des macromolécules naturelles présentes dans tous les organismes vivants. Ils sont le support du code génétique et sont, entre autres, à l’origine de la production des protéines nécessaires au bon fonctionnement du métabolisme de la cellule. Le codage de l’information génétique est assuré par l’enchainement de bases azotées (ou bases nucléiques) liées au squelette par une liaison N-glycosidique. Le squelette est formé de riboses liés entre eux par des groupements phosphodiesters au niveau du carbone C3’ d’un 1er ribose et du carbone C5’ d’un 2nd sucre, voir Figure 2-1 (E). Le ribose de l’ADN est deshydroxylé en position 2’ (voir Figure 2-1 (C)). Ainsi, les deux extrémités des séquences sont notées 5’ et 3’. Dans l’ADN, les bases azotées sont la guanine (G), l’adénine (A), la cytosine (C) et la thymine (T). Les mêmes bases sont retrouvées dans l’ARN à l’exception de la thymine qui est remplacée par l’uracile (U). Elles peuvent être classées en deux catégories : les bases puriques – les bases A et G dérivées de la purine – et les bases pyrimidiques – les bases C, T et U dérivées de la pyrimidine. La structure des bases nucléiques est donnée en Figure 2-1 (A et B). Figure 2-1 : structures chimiques des bases puriques (A) et pyrimidiques (B), des riboses et désoxyriboses (C), d’un exemple de nucléotide – l’adénosine triphosphate – (D) et d’une chaine d’ARN (E) (en bleu et en violet respectivement noyaux puriques et pyrimidiques, en rose C2’, en vert ribose et en orange groupement phosphate, entouré en rouge emplacement de la liaison N-glycosidique). Les bases nucléiques sont capables de s’apparier – c’est à dire de former des paires de bases – grâce à la création de liaisons hydrogène entre les bases. Les paires de bases classiques sont dites de type « Watson-Crick ». Ce sont les paires de bases les plus rencontrées. Il s’agit des paires G-C et A-T ou A-U dans l’ADN ou N N N H N NH2 NH N N H N O NH2 N N H NH2 O NH N H O O NH N H O O O OH OH H H H H O P O O -O N N N N NH2 P O O -O P O O --O O H OH H H H H HO OH 1’ 2’ 3’ 4’ 5’ O OH OH H H H H HO OH 1’ 2’ 3’ 4’ 5’ N N N N N H N O OH OH H H H H Base O OH O H H H H P O O O O -Base P O -O -O 3’ 5’ 3’ 5’ Purine Pyrimidine Adénine Guanine
Cytosine Thymidine Uracile
D-ribose 2’ Désoxy D-ribose Adénosine 5’ triphosphate (ATP) (A) (B) (C) (D) (E) ARN ADN
Chapitre 2 : REVUE BIBLIOGRAPHIQUE 19 l’ARN respectivement. On dit alors que les bases G et C d’une part et A et T ou U sont complémentaires. D’autres paires de bases peuvent être rencontrées comme par exemple les paires G-A ou G-U. Elles sont moins fréquentes que les premières et elles sont qualifiées de « non canoniques ». La formation de ces paires de bases au sein d’un même brin – intramoléculaires – ou entre deux séquences différentes – intermoléculaires – permet la conformation tridimensionnelle des chaines d’acides nucléiques. La conformation de l’ADN la plus connue et la plus courante dans les organismes vivants est la double hélice découverte par Watson et Crick en 1953.25 Cette double hélice – aussi appelée duplex – est créée par des appariements intermoléculaires entre deux séquences dites complémentaires – le brin 2 proposant par exemple un C en face d’un G du brin 1. Dans cette conformation, les deux brins sont antiparallèles : le premier brin est orienté dans le sens 5’à3’ et le second dans le sens inverse (3’à5’), voir Figure 2-2 (A). Les appariements intramoléculaires conduisent, eux, à la formation de structures 3D de type tige-boucle. La plus simple de ces conformations, constituée d’une seule tige, est appelée épingle à cheveux ou en anglais « hairpin » (Figure 2-2 (B)). D’autres structures, plus complexes, peuvent être rencontrées comme par exemple les G-quadruplex. Ils sont créés par la formation de liaisons hydrogène entre 4 guanines, placées dans un même plan, voir Figure 2-2 (C). Plusieurs plans peuvent se superposer et sont ainsi stabilisés par π-stacking. Les G-quadruplex peuvent être intramoléculaires ou être induits par l’interaction de 2, 3 ou 4 brins. Figure 2-2 : (A) structure secondaire et modélisation 3D d’un duplex d’ADN, (B) structure d’une hairpin et (C) exemple de structure d’un brin formant des G-quadruplex, et schéma des liaisons hydrogène formées entre les 4 guanines du plan. La stabilité de telles architectures dépend entre autres de la température. En effet, plus la température augmente et plus les liaisons entre les bases s’affaiblissent jusqu’à se rompre. On dit alors que les brins sont dénaturés. La stabilité thermique des différentes conformations peut être caractérisée par la température de fusion (Tm) qui est définie comme étant la température à laquelle la moitié des brins est dénaturée. Ainsi, plus la Tm est élevée et plus la stabilité thermique est grande. Elle augmente avec la longueur du duplex et avec le nombre de paires G-C, car ils présentent 3 liaisons hydrogène contrairement aux autres paires de bases qui n’en présentent que 2.26 La présence de sel dans le milieu permet également la stabilisation des structures 3D. En particulier, les cations divalents comme le Mg2+ et le Ca2+ ont le plus gros impact sur la stabilité, car ils sont capables d’interagir avec deux groupements phosphates pour faciliter leur
20
rapprochement et donc l’appariement des bases. Les cations monovalents ont le même effet mais dans une moindre mesure. Ils ont donc un effet positif sur la formation de duplex d’ADN ou d’ARN ou la formation de tige au sein d’une séquence.27 Les G-quadruplex, quant à eux, sont stabilisés par la présence de cations inorganiques monovalents tel que les ions K+.28 2.1.2 Définitions générales des aptamères 2.1.2.1 Quel type de cible ? Les aptamères sont des MRE qui peuvent être sélectionnés contre une grande variété de ligands. La majeure partie des aptamères décrits dans la littérature est dirigée contre des petites molécules ou contre des protéines. Les petites molécules sont caractérisées par un faible poids moléculaire – inférieur à 3 000 Da. Les petites molécules ciblées peuvent être des nucléotides, comme l’adénosine triphosphate (ATP)29 et la guanosine triphosphate (GTP)30, des acides aminés comme l’arginine31, des toxines telles que l’ochratoxine A32, des drogues (e.g. la cocaïne33), des antibiotiques tels que les tétracyclines34, ou encore des polluants organiques comme les polychlorobiphényles.35 Les macromolécules ciblées par les aptamères sont des protéines, comme la thrombine,36 la L-sélectine,37 ou des facteurs de croissance comme le PDGF.38 Les constantes de dissociation (KD) des complexes aptamère/cible sont de l’ordre du pico au nanomolaire lorsque les cibles sont des protéines et du nano au micromolaire pour les petites molécules.16 Les aptamères peuvent aussi complexer des ions inorganiques tels que le potassium39 et l’arsenic.40 Enfin, ils peuvent également cibler de plus grosses entités comme des spores (par exemple les spores de l’anthrax41), des bactéries (e.g. Escherichia Coli42), des virus comme le virus Ebola43 et même des tissus humains, généralement des tumeurs.44 2.1.2.2 Mode de sélection des aptamères : le SELEX Cette large gamme de cible découle, entre autres, du fait que la sélection des aptamères est exécutée in vitro. Cette dernière est réalisée par une méthode nommée SELEX pour « Systematic Evolution of Ligands by EXponential enrichment ».14,15 Elle se découpe en plusieurs étapes : les cycles de sélection / amplification, le clonage et le séquençage des séquences sélectionnées. Elle est réalisée à partir d’une banque contenant entre 1013 et 1018 séquences d’ADN ou ARN différentes. Ces séquences sont composées d’une partie aléatoire de 30 à 70 nucléotides (nt) de long, flanquées de deux parties constantes – le rôle de ces parties sera expliqué plus bas –, voir Figure 2-3. Lors de l’étape de sélection, cette banque est mise en présence de la cible d’intérêt. Une partie des séquences se fixe sur la cible, puis les séquences qui n’interagissent pas avec le ligand sont séparées des autres. Plusieurs techniques de séparation peuvent être utilisées : chromatographie d’affinité, filtration, électrophorèse sur gel ou plus récemment électrophorèse capillaire et l’utilisation de billes magnétiques. Les séquences affines sont ensuite séparées de la cible, s’ensuit une étape d’amplification en
Chapitre 2 : REVUE BIBLIOGRAPHIQUE 21 chaine par polymérase (PCR pour « polymerase chain reaction »). Lors de la PCR, des brins d’ADN, appelés amorces, s’hybrident sur les parties constantes des séquences pour permettre la fixation de la polymérase dont le rôle est de répliquer ces dernières. Ceci permet l’amplification des candidats aptamères pour lesquels une nouvelle étape de sélection est appliquée. Ainsi plusieurs cycles de sélection/amplification sont réalisés ; en général entre 5 et 10. Au cours de ces cycles, la population des séquences les plus affines augmente. Enfin, lorsque l’affinité des séquences restantes n’évolue plus, leur clonage et leur séquençage sont réalisés. La dernière étape est le choix de la ou des séquences aptamères parmi tous les candidats sélectionnés. Le protocole général de la méthode SELEX est schématisé en Figure 2-4. En fonction de la polymérase utilisée lors de la PCR, il est possible d’utiliser des nucléotides modifiés lors de la sélection afin d’améliorer la stabilité des aptamères en milieu complexe (voir section 2.1.2.4). Kato et al. ont ainsi sélectionné un aptamère contre la thrombine avec des 4’thio ribonucléotide (4’thio CTP et 4’ thio UTP) et des 2’NH2 pyrimidines.45 D’autres ont effectué la sélection d’un aptamère contre le facteur de croissance bFGF avec des nucléotides phosphorothiolés,46 ou encore la sélection d’un aptamère dirigé contre des cellules tumorales avec des nucléotides (5’-# BH3 phosphate).44 Figure 2-3 : construction des séquences de la banque aléatoire pour la sélection des aptamères Figure 2-4 : schéma du protocole SELEX D’autres protocoles SELEX ont été développés comme le one-step SELEX, le SELEX in vivo et le non-SELEX. Le one step-SELEX consiste en une seule étape de sélection et d’amplification. Ceci permet d’avoir une méthode plus rapide et de limiter les risques de mutation des séquences lors des étapes de PCR.47,48 Le non-SELEX est également une méthode rapide et économique. Dans ce protocole, il n’y a pas d’étape
Séquence aléatoire : 30 à 70 bases Séquences constantes pour la fixation des primers Banque de séquences aléatoires Sélection Cible Séparation des séquences non liées à la cible Élution Amplification (et transcription) Répétition du cycle 5-10 fois Dernier cycle : Clonage et séquençage
22
d’amplification entre les étapes de sélection.49 Le SELEX in vivo permet, quant à lui, la sélection in vivo d’aptamères dirigés contre des cellules tumorales.50 En effet, la complexation de la cible dépend de sa conformation qui elle-même dépend de son environnement. La sélection in vivo permet donc la sélection des aptamères contre des cibles macromoléculaires ou cellulaires dans leur état natif contrairement à la sélection in vitro dans laquelle les cibles peuvent être dénaturées. Le principal désavantage de cette méthode est dû à la faible stabilité des oligonucléotides dans les milieux biologiques. Il est donc indispensable d’utiliser des séquences stabilisées (voir Section 2.1.2.4). Enfin pour toutes méthodes SELEX, il est souvent nécessaire de réaliser une sélection négative – ou contre sélection – sur des analogues de la cible (e.g. sur des cellules saines si la cible est une cellule tumorale, ou encore sur des analogues structuraux de petites molécules cibles) et/ou sur le support de sélection afin d’éliminer toutes les séquences non spécifiques à la cible choisie.
Ainsi, plusieurs aptamères peuvent être sélectionnés pour la même cible. Le choix du mode de sélection et de la séquence in fine est régi par l’usage de l’aptamère. La séquence peut cependant être modifiée et marquée après la sélection pour s’adapter au mieux au schéma d’utilisation. En effet, les aptamères peuvent être utilisés comme outils de diagnostic, thérapeutiques et dans des méthodes de séparation – chacun de ces domaines ayant ses exigences.
2.1.2.3 Quels usages ?
Les aptamères peuvent être utilisés comme outils de détection.16 Etant des MRE versatiles et modifiables, ils peuvent être utilisés dans différents types de schémas d’analyse. Ces schémas peuvent être homogènes1 – si les sondes sont libres en solution – ou hétérogènes51 – si au moins un élément du système est immobilisé sur une surface solide. Ils peuvent également être compétitif52 ou non compétitif,53 en fonction de la présence ou non d’un compétiteur dans la solution. La nature du compétiteur varie d’une plateforme à l’autre. Il peut s’agir d’un brin partiellement complémentaire (BC) à l’aptamère4, d’un analogue structural de la cible54, d’un chromo- ou fluorophore55 ou d’une espèce électroactive.56 L’élément de transduction du signal peut être lié à une sonde57 – la sonde est alors dite marquée – ou libre en solution.55 L’aptamère peut être utilisé soit entier,58 tronqué2 ou scindé – on parle alors de split-aptamère.59 Sa séquence peut également être modifiée pour s’adapter au mieux au schéma d’analyse.18 La méthode de détection peut fonctionner en système « on »60 – la présence de la cible provoque une modification du signal – ou en système « off »61 – l’absence de la cible provoque la modification du signal. Ces schémas d’analyse peuvent être couplés à différents systèmes de transduction du signal. Ils peuvent être optiques62,63 (fluorescence, colorimétrie), électrochimiques64, radioactifs65, ou sensibles aux variations de masse51,59 (résonance des plasmons de surface – SPR – ; microbalance à cristal de quartz – QCM). Les aptamères peuvent également être employés comme sondes pour des techniques d’imagerie biomédicale.65,66 Les exemples de schémas d’analyse sont répertoriés dans le Tableau 2-1.
Chapitre 2 : REVUE BIBLIOGRAPHIQUE 23 Schéma d’analyse Méthode de détection Homo- /
hétérogène compétition marqué séquence On/Off ref
QCM / SPR Hétérogène NON NON Entière on 51,58
SPR Hétérogène NON NON Split on 59
Colorimétrie Hétérogène NON NON Split on 60
Colorimétrie Hétérogène OUI NON Entière off 61
Fluorescence Homogène OUI NON Entière off 55
Fluorescence Homogène OUI OUI Entière on 52
Fluorescence Homogène NON OUI Entière on 53
Fluorescence Homogène OUI OUI Entière on 57
Anisotropie de fluorescence
Homogène NON OUI Entière on 1
Tableau 2-1 : exemple de schémas d’analyse basés sur l’utilisation d’aptamère. Ligne rose l’aptamère, ligne bleue brin complémentaire, cible macromoléculaire (
●
), petit ligand ( ), nanoparticules d’or dispersées (●
) et agrégées (●
), graphène oxyde(hexagone noir), puce de mesure (rectangle bleu), fluorophores ( ), fluorophores éteints ( ), quencher (Q).
Les aptamères sont également employés dans plusieurs méthodes d’analyse d’échantillons complexes utilisant des techniques séparatives : la chromatographie en phase liquide (LC), l’électrochromatographie (CEC) et l’électrophorèse capillaire (CE).67 Ils ont été utilisés en premier pour l’analyse des IgE par CE.68 Dans cette étude, l’aptamère marqué par un fluorophore est incubé avec différentes concentrations d’IgE. Le mélange est ensuite injecté en CE. La mobilité électrophorétique est proportionnelle au rapport charge sur masse. Or, la formation du complexe aptamère/IgE implique essentiellement une très forte augmentation Q Q Q Q
24
de la masse ce qui provoque une augmentation du temps de migration, voir Figure 2-5 (A). Plus la concentration en IgE est élevée et plus l’aire du pic du complexe va augmenter. Plus récemment, l’analyse de mélanges racémiques de petites molécules a été réalisée.69,70 Dans ce cas, le capillaire est rempli avec un électrolyte contenant l’aptamère. Le mélange racémique est ensuite injecté. Les conditions de séparation ont été optimisées pour que l’aptamère ne migre pas. Seuls les énantiomères sont mobiles. L’ADN et l’ARN étant eux-mêmes des molécules chirales, les aptamères complexent spécifiquement la forme énantiomérique de la cible pour laquelle ils ont été sélectionnés.31 La migration de ce dernier est donc ralentie, voir Figure 2-5 (B). Les aptamères peuvent également être immobilisés sur la phase solide. La migration des solutés est effectuée par l’application d’une tension (CEC)71 ou par l’application d’une pression (LC)72. Dans ces deux techniques, la séparation est assurée par la différence d’affinité des solutés avec la phase solide stationnaire (Figure 2-5 (B)). Figure 2-5 : (A) électrophérogrammes de l’analyse d’échantillon de sérum reconstitué dopé en IgE (concentration en IgE : 0-100-400 nM de droite à gauche ; A* aptamère), (B) électrophérogramme de l’analyse d’un mélange de L- et D-arginine et (C) chromatogramme de la purification de la L-sélectine (flèche changement de la composition de la phase mobile pour l’élution de la protéine cible). Adaptée des références 68–70,72 Enfin, l’utilisation des aptamères à des fins thérapeutiques est de plus en plus décrite.73 Pour l’heure, un seul aptamère a reçu une autorisation de mise sur le marché par la FDA (« Food and Drug Administration »). Il s’agit du pegaptanib dont la forme pharmaceutique est connue sous le nom MACUGEN. C’est un aptamère dirigé contre le facteur de croissance VEGF impliqué dans la dégénérescence maculaire liée à l’âge (DMLA).21,74 Deux autres aptamères sont actuellement en phase III des essais cliniques : le pegnivaogin qui pourrait être utilisé comme anticoagulant, et le pegpluranib pour traiter la DMLA via l’inhibition d’un autre facteur de croissance, le PDGF.74 D’autres études utilisent les aptamères comme outils de vectorisation afin d’adresser spécifiquement des principes actifs vers leur cible.75 Par exemple, Chen et al. ont modifié des nanoparticules avec des aptamères dirigées contre le noyau de cellules tumorales afin d’y libérer spécifiquement un anti-cancéreux – la doxorubicine. L’avantage des aptamères par rapport aux AC pour les applications thérapeutiques est qu’ils sont non immunogéniques.13,76 Cependant les aptamères ayant un poids moléculaire faible – entre 8 000 et 15 000 Da –, ils sont facilement éliminés par l’organisme par voie rénale. Pour améliorer leur biodisponibilité, leur taille peut être augmentée en les liant à un motif polyéthylène glycol (PEG), un liposome ou autres.76 De plus, leur utilisation est limitée par leur faible stabilité
nM. Using a previously described model for competitive bind-ing,5,29we estimated the K
dof unlabeled aptamer-IgE to be 8 nM from the competitive calibration data. This calculated value agrees well with the published value of 10 nM.26 Based on these numbers, it is apparent that addition of fluorescent label caused a significant decrease in binding affinity of aptamer for IgE. This is not unexpected as the tertiary structure of the oligonucleotide may be dependent upon the access to both the 5′ and 3′ ends for interaction. Other labels, linkers, or label positions could be used to optimize labeling and minimize the effect on binding.
Complex Samples . The possibility of performing APCE analysis in complex mixtures containing high concentrations of potential interferences was assessed by carrying out the assay in human serum. In this experiment, samples of human serum spiked with various concentrations of IgE were mixed with A* to a final concentration of 300 nM and analyzed. Figure 5 shows a series of electropherograms from different concentrations of IgE for this experiment. The results show that the complex sample had little effect on separation, dynamic range, or detection limit for the assay. The only observable effect was an increase in tailing of the A* peak in the presence of serum (see expanded portion of Figure 5). This effect may be due to weak binding of A* with some substances in the sample or an injection artifact caused by the serum sample. This experiment demonstrates the ability of aptamers to selectively bind to their target in the presence of the large amount of interferences and the possibility of performing clinically relevant assays on biological fluid samples.
Aptamer-Bas ed AP CE for Thrombin. The principle of aptamer-based APCE was further tested using a thrombin aptamer to detect thrombin. Thrombin aptamer is a significantly weaker binder than IgE aptamer with a Kdof∼200 nM.27Figure 6A shows a pair of electropherograms obtained under conditions of no flow in the capillary. The electropherogram on the right shows that in the presence of thrombin the A* peak decreases and a large tail to the right of the A* appears. The tail has a fairly abrupt end at∼50 s, indicative of a complex peak that is rapidly dissociating during the separation causing bridging between the zones for free and bound A*. Raising the pH to 8.2 and application of vacuum-induced flow as done for the IgE assay yields a reasonable complex peak as shown in Figure 6B. Even with these
conditions, however, the baseline separation of the complex from the free aptamer was not possible due to the bridging effect.
A calibration curve for thrombin based on peak heights had characteristics similar to that for IgE. The detection limit based on detection of the complex peak was calculated to be 40 nM. The higher detection limit relative to that obtained for IgE was presumably due to more significant loss of the complex by dissociation resulting from the poor binding constant. A competi-tion experiment between labeled and unlabeled thrombin aptamer revealed specific binding by the A*. On the basis of the curves obtained from that experiment and the calibration, we measured dissociation constants for both A* and unlabeled aptamer to thrombin to be∼450 nM which is ∼55%higher than the published value of 200 nM.27 This discrepancy may be due to use of incubation conditions different from those used for selection, or perhaps rapid dissociation during the separation may have caused the dissociation constant to appear higher. Interestingly, both A* and unlabeled aptamer had similar dissociation constants, suggesting no effect of the label on binding for this aptamer. CONCLUSION
The data presented here demonstrate the feasibility of using aptamers as affinity probes in noncompetitive APCE assays. Use of conditions that promote rapid separation and migration allows observation of both free and bound aptamers even for dissociation constants of hundreds of nanomolar. Detection limits, linearity, selectivity, and assay speed are all suggestive of assays that could
Figure 5 . Determination of IgE in serum. Electropherograms obtained for samples prepared in reconstituted human serum. Each sample contained a final concentration of 300 nM A* and 0, 100, and 400 nM IgE in A-C, respectively. Inset demonstrates a high-sensitivity analysis of a sample containing 0 and 5 nM IgE with 300 nM A*. Inset is at 10-fold higher scale than the other electropherograms.
Figure 6 . Determination of thrombin using FITC-labeled aptamer. (A) Electropherograms obtained for 2000 nM A* with 0 (left) and 3000 (right) nM thrombin at pH 7.4 and no flow. Fluorescein (50 nM) was used as internal standard (IS). A large tail next to the free A* indicates the presence of the quickly dissociating complex. (B) Electrophero-grams obtained for same concentrations (50 nM 4(5)-carboxyfluo-rescein as IS) at pH 8.2 and application of vacuum.
4544 Analytical Chemistry, Vol. 70, No. 21, November 1, 1998
(A) (B)
T.S.Romig et al./ J.Chromatogr.B731 (1999) 275–284 279
Fig. 1. AAC of partially purified LS-Rg fractions by multiple gentle elution methods. For panels A, B and C, approximately 220 mg of total protein was loaded on a 1 ml AAC column at a volumetric flow-rate of 0.75 ml / min. After washing the column with PBS until baseline was achieved, LS-Rg was eluted from the column by a linear EDTA gradient (A), a linear NaCl gradient containing no divalent cations (B) or containing 1 mM calcium and magnesium (C). Panel (D) is the same as (A) except that approximately 1.2 mg of total protein was loaded on the column. Because EDTA absorbs weakly at 280 nm, a slight rise in the baseline was observed for these elution profiles. Arrows indicate the initiation of the gradients.
and protein blot experiments, it was possible to epitope and that the LS-Rg was highly purified (Fig. analyze the fractions for each of the three purifica- 2C). The band with the slowest mobility was the size tion trials. By protein blot analysis, no LS-Rg was expected for the glycosylated LS-Rg dimer. The detected in the column flow through for any of the identity of the other two bands is uncertain, although three purification trials (Fig. 2C) while the g- the one with the greater mobility may be the globulin fraction was separated (Fig. 2A). As de- monomer.
termined by silver stain analyses of SDS-PAGE Similar results were obtained by protein blot experiments, the elution fractions for all three con- analysis using a novel detection system for nonnu-ditions were identical and consisted of three distinct cleic acid binding proteins [32]. In this case a bands (Fig. 2B). All three protein bands for each standard protein blot procedure was utilized (see elution condition were recognized by anL-selectin Experimental) except that the detection probe was specific antibody (DREG56) in protein blot experi- the same biotinylated aptamer as used for AAC. A ments indicating that all three bands share a common streptavidin-linked AP detection system was used to
L-Sélectine
Matrice (C)
Chapitre 2 : REVUE BIBLIOGRAPHIQUE
25
dans les milieux complexes en raison notamment de la digestion enzymatique des séquences par les nucléases. Leur demi-vie dans le plasma humain est de quelques secondes pour l’ARN et de 30 à 60 min pour l’ADN.76 Afin de limiter ce phénomène, les séquences peuvent être modifiées chimiquement. Il est également possible d’utiliser des spiegelmers. Ceci est détaillé dans la section suivante. 2.1.2.4 Stabilisation des acides nucléiques L’utilisation des aptamères en milieu complexe est limitée par la digestion enzymatique des séquences. Pour pallier ce problème, la structure des oligonucléotides peut être modifiée : au niveau du ribose, des phosphates ou des bases. Dans le cas de la modification du ribose, le carbone C2’ peut être substitué par un nombre important de groupements : Fluor (-F), méthoxy (-OMe), amine (-NH2), thiol (-SH), azide (-N3), hydroxyméthyle (-CH3OH), voir Figure 2-6. Ce type de modification permet d’augmenter la demi-vie de l’oligonucléotide au-delà de 81 heures en fonction du groupement placé en position 2’. L’introduction d’un pont entre l’oxygène en 2’ et le carbone 4’ améliore également la résistance à la digestion enzymatique. Ces nucléotides sont appelés Bridged Nucleic Acids (BNA). Ce pont, qui fige la structure du sucre, peut être un simple méthyle, on parle alors de Locked Nucleic Acids (LNA), un aminométhyl (-CH2NH-), un méthoxy (-CH2 O-) ou encore des structures plus complexes comme (-CH2O CH2(Ph)-). Leur structure est donnée en Figure 2-6. A l’opposé, les Unlock Nucleic Acids (UNA) permettent une très grande flexibilité du sucre en rompant la liaison C2’-C3’ (voir Figure 2-6). Ils augmentent eux aussi la résistance aux nucléases mais sont plus simples à synthétiser que les LNA.20,21 Kato et al. ont, quant à eux, modifié le sucre en remplaçant l’oxygène du furanose par un atome de soufre. On parle alors de 4’ thio ribonucléoside. Il possède une meilleure stabilité thermique que son analogue oxygéné et résiste mieux à l’attaque des nucléases.45 La première modification à avoir été utilisée pour augmenter la stabilité des séquences d’oligonucléotides a été le remplacement d’un oxygène en # du phosphore du nucléotide par un soufre. On parle alors de (5’-# thio phosphate). Bien que cette modification améliore la stabilité des oligonucléotides, elle entraine également une forte adhérence aux protéines.20 L’oxygène en # du phosphore peut également être remplacé par un groupe borane (5’-# BH 3 phosphate).21 Ces séquences peuvent ensuite être utilisées en radiothérapie si on utilise l’isotope radioactif du bore (11B). 44 La stratégie la plus simple est d’effectuer les modifications après la sélection de l’aptamère, mais ceci entraine une diminution voire une perte totale de l’affinité si elles affectent le(s) site(s) de fixation de la cible. Il est alors possible de ne modifier que les extrémités des séquences, ce qui permet une protection contre l’attaque des exonucléases. Cependant, il a été montré que la modification des tiges, qui n’interviennent pas dans la reconnaissance, peut tout de même affecter l’affinité de l’aptamère pour sa cible.20 Une autre stratégie est d’introduire ces modifications pendant la sélection par l’utilisation de nucléotides modifiés lors