Detection of long repeat expansions from PCR-free whole-genome sequence data
Texte intégral
Figure
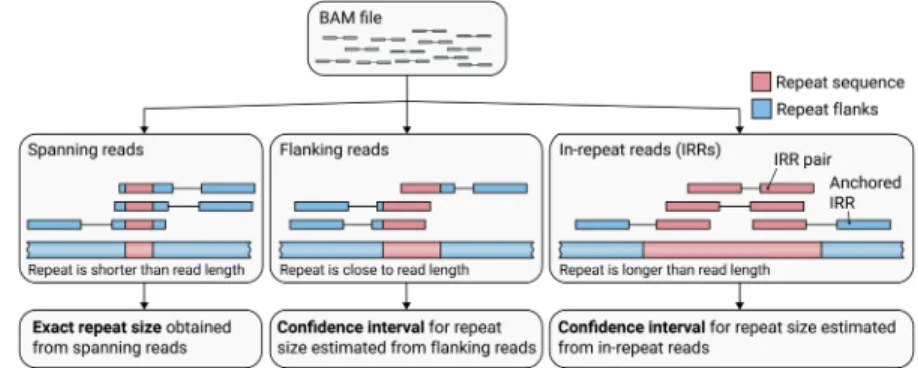
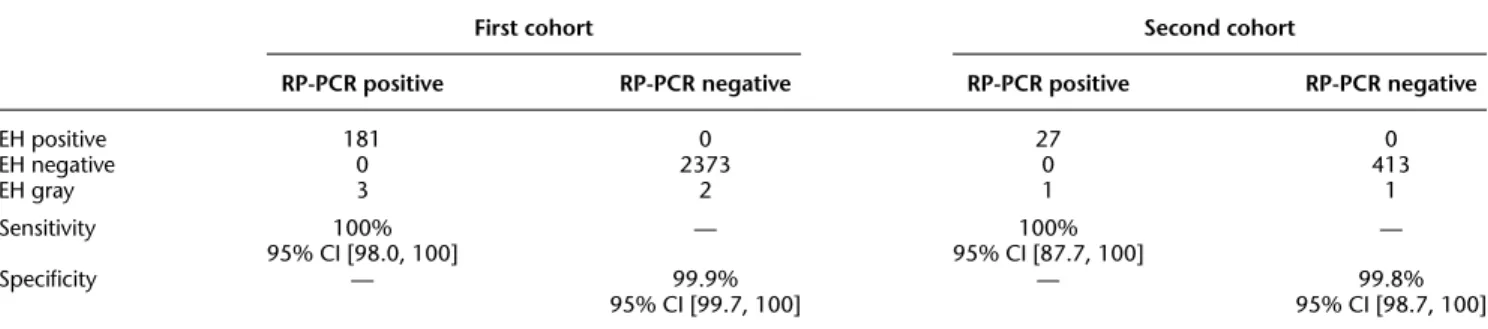
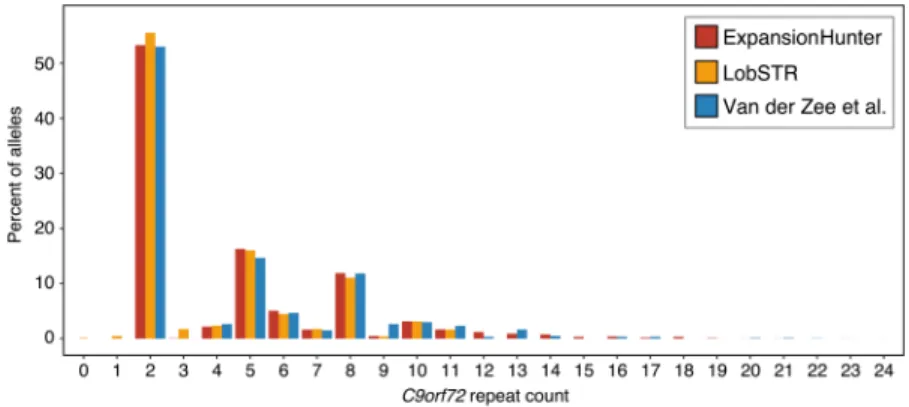
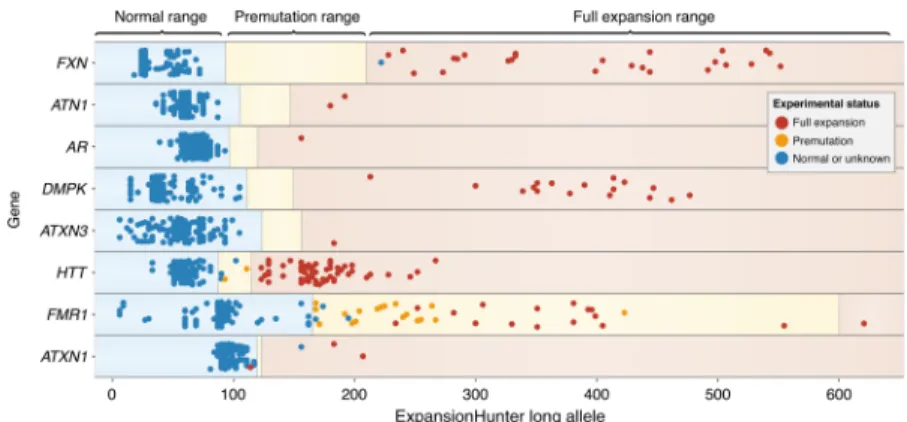
Documents relatifs
(ii) aligned these reads back onto the reference genome, using the same parameters as used for the experimental HTS data; (iii) identi- fied the chromosomal coordinates of the unique
Local minima were obtained by performing a zero-temperature Monte-Carlo simulation with the energy function in Eq (2), starting from initial conditions corresponding to
In theory, similar or better prediction accuracies than those reported in this study can be obtained if data on all the individuals in the meta-GWAS are available, and if they
Background: This study aimed at (1) comparing the accuracies of genomic prediction for parasite resistance in sheep based on whole-genome sequence (WGS) data to those based on 50k
In the impcons strategy that targets both genetic improvement and conserva- tion, using estimated relationships based on SNP chip or WGS data resulted, respectively, in the
Summary statistics of Breakdancer (BD), Pin- del (PD) and Overlap method in HOM and HET simulation sets; Table S3: Precision of each SV detection method for deletions, inversions
Functional enrichment analysis of genes that are spe- cific to the YNLC breed revealed that many candidate genes are related to immunity and disease resistance (see Additional file
Descartes’ rule of signs and the identifiability of population demographic models from genomic variation data.. Efficient inference of
