ANIWiRSITE
8MAI45
GUELMA
ffi
Facultd
de
mathdmatiques
et
informatique
et
dessciences
de
la murtiire
Ddpartement sciences
de
la
mutibe
Mdmoire
en vae
de
l'obtetntion da
dipbme
deMaster
en
physique
REPUBLIQUE
ALG]ERIENNE
DEMOCRATIQT]E ET
POPULAIR]E
MINISTERE
DE
L'ENSEIGNEMENT
SUPERIEUR
ET
DE
TA RECHERCHf, SCM,NTIFIQUE
Prdsentd
pat :
Hamici
Selma
Sous
la
direction
de
Dr.S.
Djeroud
,'Aed
.
nr[.y,
ok*-,t*l
l]l"l
Les
Prollridtds structurales et 6lectroniques
des
composds
i
base
des
6ldments
des
groupes
lllA
et
VA
Remerciements
Un
trds
grond
merci:,
au
bon
DIEU
qui
est
toujours prdsent
auec
moi dans
le
bonheur
et
dans
le
pire.
Je tiens
d,remercier
uiuem€rlt
(M,ne
.Djeroud
r
maitre
de
conf4rences
d,l'uniuersitd
de
Guelma d'auoir
prapos6,
dirig6
et s;uiui
ce
trauail.
J'exprime toute
m,a
reconnaissq,nce
d,Monsieur B.
Benneccer
professeur d,l'uniuersitd
ale
Guelrna
et
Dr
F.kalarasse
dhuoir
accetptd
d'examiner
ce
trauail.
Je
tiens
4galement
d
rernercier
M"'B.Belfarhi,
Manl,tre
d,econfdrences
d
I
uniuersit:d
de
Guelma,
qui
a
bien uoulu
m,e
faire
lh,onneur
d'ernminer
ce
trauail
et
de
pr6sider
Iejury.
Ma
gratitud,e
d,
uDr
Larnia
Kalarasse>> que
je
considdre
commle
tna
grande
s@ur
toute
en
lui
souhaitant une
bonne rdussite
dia;ns
sa
carridre scientifique.
Je tiens
d,remercier
tous
ceux
qui
mbnt
aid6e
de
prds ou
de
la,in,
scrns
oublier tous
les
mem:,bres
du
laboratoire
(
LPG
t
paur
leur
ai:de
taut
au
long
de
la
rdalisailion
de
ce
trauail.
J*
I
R6sum6
Le
but de
cetravail
estd'dtudier
les propridtds structurelleset
dlectroniques
ii
base deselements des groupes
IIIA
et
V\
cristallisant dans la structure zinc-blende(BN,
l\ll',
GaAs,AlN, AlAs et
GaN),par
la
methode des ondes planes augmentees linearisees (F.P-JLAPW),dans
le
cadre dela
theorie de la. fonctionnelle dela
densitdDFT,
impl€mentee dansle
codeWien2K.
Nous avons
utilis6
I'approximatjonde la
densitelocale
LDA, pour le
terme dtr
potentield'echange
et
de
corrdlation(XC),
pour calculer les
proprietes structurales(pas
Ce rdseau,module de
rigidite)
bien que lesproprietes
electroniques (les structures de bandes, nature dfiLes r6sultats obtenus montrent que
la
structure zinc-blende presente un gap direct iru pointf
f-.-pour (GaAs, GaN) et un
gap
indirect
f-X
pour
(BN,AlP, AIN
et AIAs).On a trouvd que le gap
diminue
lorsqu'on descend dans les colonnes des groupesIII
etV
etceci est d0 certainement d I'augrnentation du nombre
atomiqueZ.
Nos
r6sultats obtenussont en
bon
accord avecles
donndes expdrimentaleset
l,es calculs thdoriques.Abstract
The aim
of
this work isto
study the structural, electronic databaseof elements groups ofIIIA
andVA
crystallizing in zincblendestructure (BN,AlP,
GaAs,AlN,AlAs
andGaN).
Ey
themethod of linearized augmented plane wave (FP-Lapw).under the theory of density
functional DFT, implemented
inthe
codeWIEN2k.We
used
the localdensity
approximationLDA
for
the termof
thepotential for exchange and correlation (XC), to calculate structural properties (no network, stiffiress), althoughtheelectronic prcperties
ftand
structuues, nature of the gap and thetctal
densitiesof
statr:serndpartial).
The :results show that zinc-blende structure has a
direct
gap atfpoint
for (GaAs,CrN)
and an indirect gapf-X
(BN, AlP,
Allrl
andAlAs).It
was found that when the gap down in the columns of groupsIII
andV and this iscertilinly
the increasing atomic number Z.
Our
results obtained arein
good
agreementwith
experimental
dataandtheoreticalaa
Somm,aire
Sommaire
Introduction
96n6rale
Chapitre
I
Th6orie
dlela Fonctionnelle
de
la Densit6
(DFT)
Introduction...
...'...'3i.l.Equation
deSchrodinger.
...3
L 2..L'Haniiltonien
total ducriscal
...'.'.3l.
3 .L'approximation de Born-
tlrppenheimer..
...4I. 4 .L'approximation de
Hartree-Fock.
...
...5I.
5. Lathdorie de la Fonctionnelle de la Densit6(DFT)
...
...7I.S.l.Les thdordmes de Hohenberg et
Kohn.
...7
I.5.2. Les dquations de Kohn-
Sham
...8L
6.L'Approximation
de la Densitr! locale(LDA)
' '. '. ' ...'9I. 7.L'Approximation
du Gradient Gd,ndralisd(GGA)
...10I.
8 Solution de l'dqr.ration de Kohn ctSham
-'....
...10I.9. L'auto-coh6rence dans les
cal:uls.
" " "
""1
1Chapitre
II
M6thodes
de rcalcul
des
structures
de
bandes
6lectroni.ques
Introcluction...
""""""1r
ILl.
Mdthodes de calcul6ldmentaires....'.
""""""14
II.1.1. M6thode de calcul des dlectronslibres.
" "
'
"
"
'14II.I.2.
M6thode des dlectrons presque libres(N.F'E.M).
'
"""14
II.2.
Methodes de calculavanc6es.
""
'i5
li.2.I
Lamdthode des liaisons fcrrtes(L.C.A.O).
" ''
"' 15Il.3.lMdthocles des ondes
planes.
"""""'16
II.3.1.Mdthocle des ondes planes
orthogonalisdes.
"'""""16
II.3|.2. La mdthode des
pseudopotr:ntiels
"
'"
""17
Il.4.Autres
mdthodes.
" "
"""
18Il.4.l.Mdtliode
cellulaire...'
""
"""18
Il.4,2,Mdthode des ondes plarresaugmentees.'....
''""18
Sommaire
Chapitre
III
la
m6thocle
desondes planes augment6es lin6aris6es
(FP-LAPW)
Introrjuction
...
...2(lIIL l. La mdthode des ondes planes augment6es
(APW)
...,....21IIl.2.La
rndthode de ondes plarres augmentdes lindaris6es(FP-LAPW)
...
... .. ...2:\IIL.2.I.Les bases de la m6thcde
(FP-LAPW)
.
.. ...23III.3.Les rdles des dnergies de linriarisation
El
.......24
Ill.4.Constructions des fonctions
rlcliales
...2t''IIL4.1.
Les for-rctions radiales nonrelativistes
...,...25IIL.4.2.Les foncions radiales
relativistes
... ...26ili.S.Resolution de I'equation de
Poisson
..'....28
Ill.6.Amdlioration
de la rndthode(FP-LAPW)
.
..., . , ....29III.6.l.Les
fendtres d'energienrulti;:les
,. ...29Ill6.2.Developpement
en orbitalelocale
...'..30Ill.7.'fraiternent
des efl'ets de spin-orbite....
. '....30III.8,
WienZk...,...
, .;..31Chapitre
IV
lR6sultats et discussion
IV.l.Ddtails
decalculs
.."...34IV.i.l
Testdeconvergence...,
...37IV.2.Propri6tds
structurale...
...'.46IV.3.Les proprietds
6lectroniques.,...
...52Iv.1.3.l.Structure
de bandesd'6nergie.
'...521Y.4. Ladensit6 d'6tats
(DOS).
....'...58
V.
Conclusion g6n6rale...
...'...."""67
Sommaire
Liste
des
figures
Figures
Titres
pages
Fig. Diagramme Cycle auto
coh6rent
de lath6orie
dela
fonctionnellede densit6 (DFT)
t2
Fig.
II.
1 Syntliese d'une onde p,lane orthogonalisde:
(a) Onde plane(b) Fonction d'onde du cceur atompique. ( c) Onde plane orthogonalisee
17
Fig,lll.
I Potentielcristallin d'un
rtlseau carre d deux dimensions:(a)
potentiel total, et (b) potentielmuffin-tin.
2Q
Fig.lll.2
Potentiel <Muffin-Tirr
>(MT)
21Fig.lll.
3 Les fendtres d'dnergie multiPle 5UFig
.lll.4
La structure du programme Wien2k 33Firr
lv.
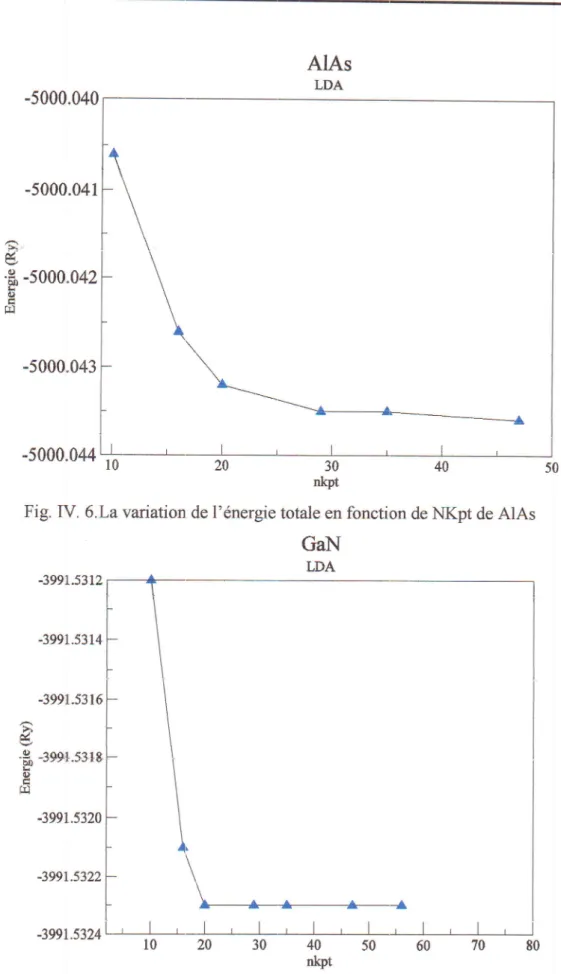
1 Structure zinc-blende35
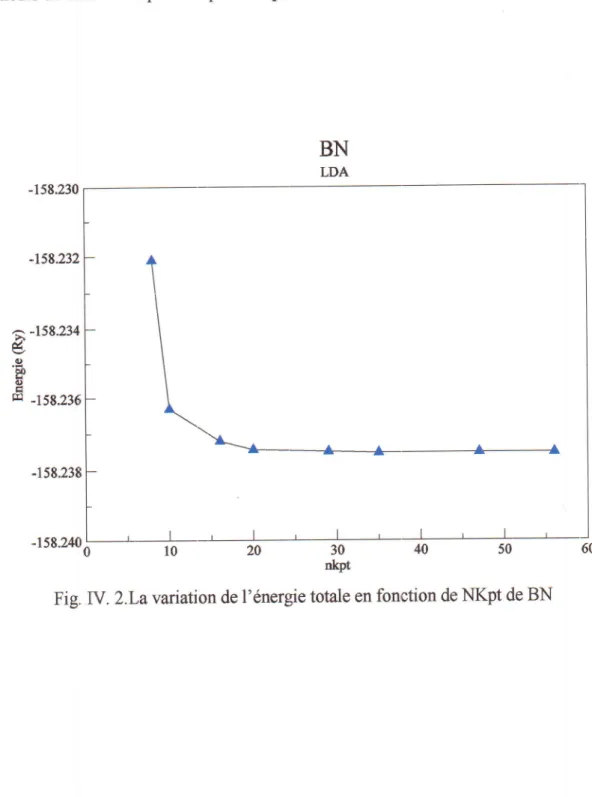
Fig. IV.2 La
"arrtton
de l'6nergie totale en fonctionde
NKpt
deBN
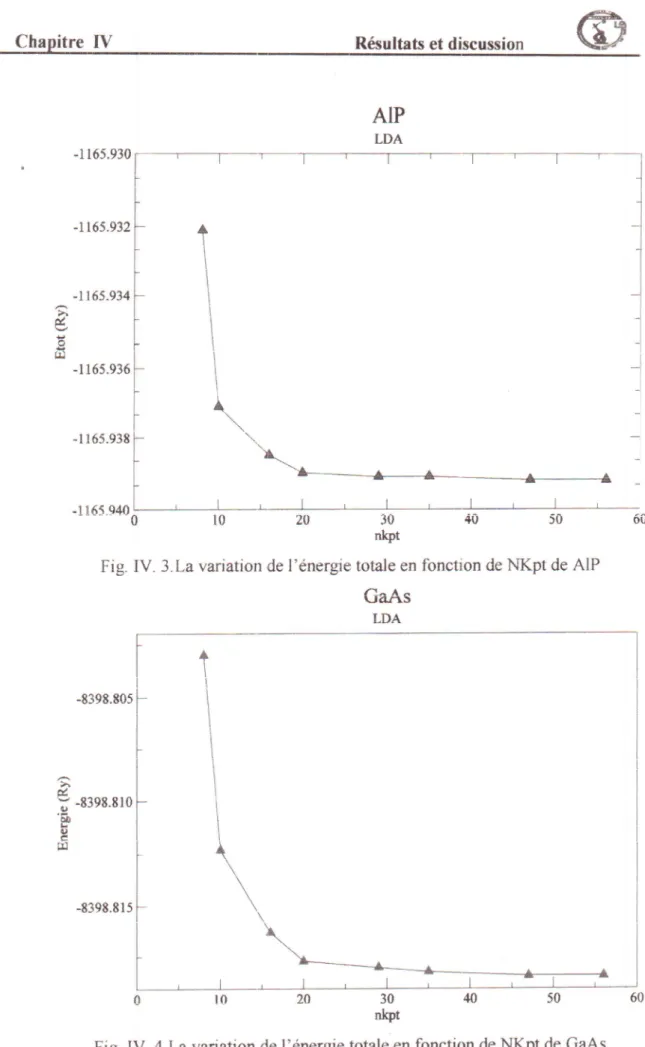
f.a variation de l'dnergie totale en tbnction deNKpt
de AIPL"
"t'iatitn
de l'dnergie totale en fonction deNKpt
de GaAs37 38 Ilirt ''b'
lv.
J 38 Fig.IV.4
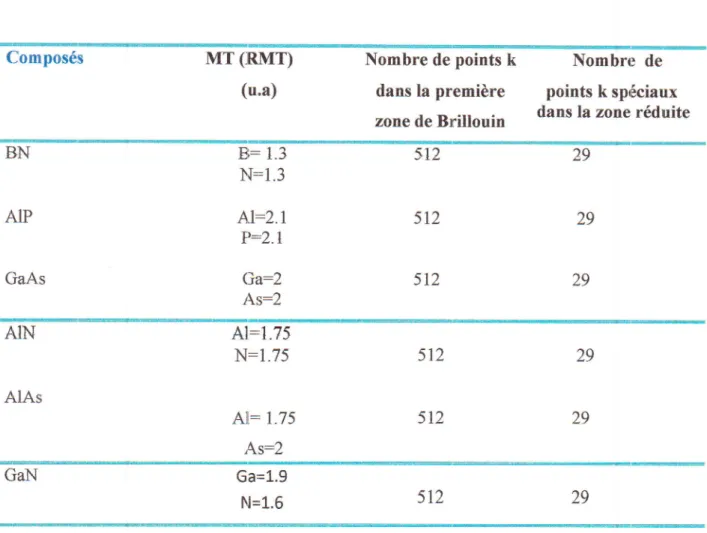
Fig. IV.5
L"
*t*tro"
dt'dnergie
totaie en fonction deNKpt
deAIN
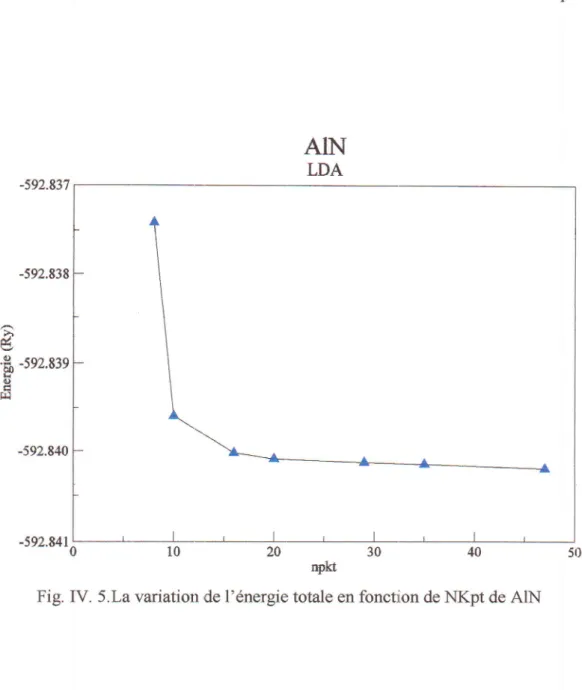
ffig*to
ffijie
totale en fonction deNKpt
de GaNLa variation de I enerlgie totale en fonction
de
Rkmax de BNI-a variation cle l'6nergie totale en fonctiou de RKmax de AIP
-L^*i.t.'t
de l'6nergie totale en fonction de RKniax de GaAs
f_u
u-iatio"
aet
etre-gie totule en fonction de RKmax deAIN
39 40 Fig.
lV.6
40 Fig.iV.7
A1 -L Fig. IV.8 A' +-) Fig. tV.e 43 Fig.IV.l0
44 Fig.IV
.l
ISommaire
Fig. IV.12. La variation de I'6nersie totale en fonction de RKrnax de AlAs 44
Fig.
IV
1aIJ La variation del'6nersir'totale
en fonction de RKmax de GaN 45fig.
IV.14. Variation de 1 'dnergir: totale deBN
(Zinc blende) en fonction du volume alA-fFig.
IV.l5
Variation deI
'6nergie totale de AIP (Zinc blende) en fonction du volume atA1Fig. IV.16 Variation de 1'6nergie totale de GaAs (Zinc blende) en fonction 48
Fig.
IV
17 Variation de I'energie totale deAIN
(Zinc blende) en fonction 48Fig.
IV.l8
Variation deI
'6nergie totale deAlAs
(Zinc blende) en fonction du volunte 49Fig. IV
l9
Variation deI
'6nergie totale de GaN (Zinc blende) en fonction du volume 49Fig. IV 20 La structure de bandel; de
BN
gap indirecte en phase zinc blende 52Fig. IV
2I
G
iiucture
de bandes de AIP gap indirect en phase zinc blende 53Fig.
n/
22 Lu rtr-,r.tute de bandes de GaAs gap indirecte en phase zinc blende 53Fig
I\/.23
ta
structure de bandes deAIN
gap indirecte en phase zinc blende 54Fig.LY.24
L"
rtrr-*t*'e
cle ban,ies deAlAs
gap indirecte en phase zinc blendeL.
rtr,"t."ee
bandcs de GaN gap directe en phase zinc blende'ffiT.
ffi-p'
@
ffi-potti.tt"
a"
AIP (Zinc blende)ffi@
54 55 F'i rl ^'b' I\/lv 25 58Fig.IY
26 59Fig.I\,/
27 60llio
^ ^b'lv.
28 61 lli,rIV.
29 62 F'i o - 't' IV. 30Sommaire
Fig.
IV.31
Densin6 d'etats (DOS) f,artidlle de GaAs (Zinc blende) 63Fig.
IV.32
Densit6 d'dtats (DOS)totale
deAIN
(Zinc blende) 64Fig.
IV
33 Densitd d'6tats (DOS) totale deAlAs
(Zinc blende) 65Sommaire
Listp
des
tableaux
Titres
ffif
dilBfAP,AlN,AlAs
et GaN utilis6s dans noscalculs
par
la mdtirode FP-LAPWd6rivde
B'.
L'orreur
relative de paranldtre du r6seau.Valleurs des 6nergies des
calculdes avec la
LDA
ainterdites de la phase zinc blende
x points sp6ciaux .
IV.5
Differents niveaux tinerg tique et paramdtre de rdseaux.IV.4
5657
t*
Introduction
g6n6rale
I
ntrotl
u
ction
gd ne
ral
e
La
physique des
mat6riauxjoue un
rdle de plus en plus
important dans
lesapplications technologiques, et ce r6le ne fera que progresser dans beaucoup de domairres.
[l]
Vu
l'dpanouissement
de
la
tschnologie,
de
I'information
et
les
besoins
deI'industrie,
les composds semi-conducteursIII-V
cristallisant dans la structureZinc
trlende ont6td
un
sujet
de
speculationet
extrapolation,en particulier
d
causede
leurs
appliLcationspotentielles
dans
la
r6alisation
et
le
d6veloppement
des
dispositifs optiques
etoptodlectroniques. [2]
Pour comprendre les dilferentes propri6t6s des solides,
il
est ndcessaired'$tgdiel
lecomportement des dlectrons dans ces derniers, et ceci est en
lien
direct avec les me'hodes decalcul
qu'on
classe entois
groupes:
Les
mdthodes empiriques, semi-empiriquesr ertenfin
celles
dites ab-initio,
basdessur
la
thdorie
quantique fondamentale,utilisent
seulement lesconstantes
atomiques
comme
paramdtresd'entr6e pour
la
r6solution
de
l'dqrntion
de Schrodinger. Ces m6thodessont
devenuesaujourd'hui un
outil
de
basepour
I'r:tuLde clespropri6tds
structurales, 6lectroniques, mf,saniques,
optiques,...
des
moldcules
et
desmatdriaux.
Elles
sont
aussiun
o,util de
choix pour
l'dtude de
certainseffets
difliciles
ouimpossibles
de
ddterminer
par voie
experimentaleet
pour
la
prddiction
de
llouveau(
mat6riaux,
et
elles
ont
parfois
pu
remplacer
des
exp6riencestrds
co0teusesc,u
m6me irr6alisables au laboratoire.La
puissance des calculs ab-initio a pour origrne le forrnalisme de la thdorie de lafonctionnelle
dela
densitd(DFT)
et
ses deux approximations del'dnergie
d'6chalrg,eet
decorrdlation : I'approximation de la densitd locale
(LDA)
et l'approximation du gradientgdn€ralisd (GGA). Le formalisme de base de la DFT est bas€ sur le thdordme de Hohr:nlberg et
Kohn (1964), qui repose sur la corrsiddration que l'dnergie totale
d'un
systdme est unr:fonctionnelle de la densit6 6lectrorrique. [3,4]
Parmi
les
m6thodes ab-irLitio, 1a m6thode
FP-LAPW
(full
potential
-Lintanzed
augmented plane wave) est
l'une
des plus pr6cises, actuellement, pour le calcul de la structuredlectronique des solides dans
le
c,adre dela
thdorie dela
fonctionnelle dela
densitd (DFT). Cette thdse a pour but de contribu,eri
la ddtermination des propri6t6s structurales etIntroduction
g6n6rale
dlectroniques des compos6s d base des dl6ments des groupes
IIIA
et
VAo en utilisant lamdthode des ondes planes augmentdes lin6aris6es
(Fp-LApw).
Nous avons 6tudi6, en
particulier
lescompsds
binaires
BN,
Alp,
GaAs,AlN, AlAs
et GaNde la
famille
des semi-conducteurstrI-V
se
cristallisant dans la structure zinc-blende.Le
tavail
que nous prdsentons dans ce mdmoire comprend deux parties:La
premidre partie est consiacrde aux fondements th6oriques, elle est composde de troischapitres.
Le
premier est
destirLdau
fondement
de
la
th6orie
de
la
fonctionnelh de
la
densit6(DFT),
I'approximation
de
la
densitdlocale
(LDA)
et
l'approximation
du
Epadient
gdndralisd
(GGA)-
Le
deuxidme prdsente quelques mdthodesde calcul
des struct*res
debandes dlectroniques et
le
troisid:me, chapitre est consacrd d 1a m6thode deFp-LAp.W
(full
potentiel Linearized
augmented .planewave)
que
nous avonsutilis6e
afin de ciilculer
la
structure de bandes dlectroniques.
La
deuxidme
partie
rdsume
nos
rdsultats,
leurs
interpretations
ainsi
qu,unecomparaison avec certains
travarx
th6oriqueset
expdrimentaux disponiblesen
littdrature.Dans cette partie, on trouve l'6fude des propri6t6s structurales des compos6s
B\
Alp,
GaAs,AlN, AlAs
etGalri
et le calcul des propri6t6s 6lectroniques de ces mat6riaux.Finalement, notre
travail
est achev6 par une conclusion gdndrale rdsumant nos r6sullats les plus saillants.Introduction
rfn6rale
Bibliographie
i
[1]
H.Mathiew, Phisique des semi-condqcteurs et des composartts dlectroniques, Ed..Miasson(1 ee8).
[2]
Gj.ackland, rep.prog.phys. 64 (2001D 483 and references cited therein.tslP. Hohenberg and
W.
Kohn, Phys. Rgv.B
136 (1964) 864.Chapitre
I
ft-/€h,
Chapitre
I
Theorie
de lafonctionnelte
de
la
densit6(Dr r
r .\ry
J__
L
Th6orie
dela fonctionnelle
dela
densit6(DFT)
Introduction
Les
propridtds physiquesd'un
solide sont extrcmement lides au comportementdes dlectrons qui le constituerrt. Pour une comprdhension fondarnentale de
la stnlcl;ure
dlectronique
et
des propridtesr des matdriaux,le
ddveloppement dela
thdorie
de lafonctionnelle de
la
densitdDIrr
avec les approximationsLDA
ertGGA
ajoue
urr r.oletuds important dans la
physique
de la matidre condensde. [a]Ll.Equation
deSchriidinger
un
corpscristallin
est un ensemble de noyaux et des electrons eninteraction. En 1926'
le
physicienAutrichien
schrodinger a proposdune equartion
qui
ddcrit
t'utes
ces interactions
La
rdsolution de l'6quation de schrodi ngercaractlisant
ce sys1,,nepermetfrait de d6crire ses propridGs quantiques et elle
est donnee par :
[l]
Hw:
E\r
(r.t)
of
E
:
estl'dnergie
totaredu
systdme etyfr,
Fo)
est rafonction
d'onde, et E[ e;stl'Hamiltonien
de ce systdme, pour un systdme ayantN noyaux etrn 6lectrons.
f.2.L'Hamiltonien total
du
cristal
L'6tude
des structures cle bandes perrnetd'interpreter
phnsieurs ph6nomd;nesphysiques, qui se ddroulent dan;s les colps solides. Dans ces corps., cette structure peut
6tre obtenue en tenant compte de toutes les interactions existant
enttre les noyaux et les
electrons'
Les
r6solutions des 6quations g6ndralessont
impossibles,mais
on
per$toujours adopter
des
moddles simplifi6s
pour
pouvoir
obtrsnir
des
solutions
approchees.
Il]
L'Hamiltonien
total est ddfinipar:
H:
T,
+
To+Vo
*Ven *Vnn
T
e
repr6sente l'dnergie cindtique des dlectrons.Chapitre
I
-
'fh6orie
de lafonctionnelle
de ta densit6(I)
-'.-'-'*'-'
'
DFT)
'(P
T
n
I'dnergie cin6tique des noyaux.V""
I'dnergie d'atffaction 6lectron _ dlectron. Ven
l'dnergie d'athaction dlectron _ noyau. V oo I'dnergie d'attraction ru'yau _ noyau.L'Hamiltonien s'ecrit
n=fiZtvr,
+
t/zZT
zl*o#zT
EI
ffi
-
rTEI #*v
n, +
L/zzyzyffi
(r 3)
Au:
m:
est La masse de l,6lectron.Fii:
estla distance entre l,6lectroni
et l,6lectron7.fi
:
estla masse du noyau.Rs:
est la distance enfie les centres des noyaux k er/
Zy ,21: Les nombres atomique
s
des noyaux k etI
Les
diverses mdthodes
de
roalculde
la
structure
de
bandes dlectroniquesdtes
matdriaux iLL'e1at solide miser; au polnt
au cours des denridres ddcennies reposent sur
un certiain nombre d'approximertions reparties sur trois niveaux :
fJ.
Approximation
deBorn-roppenheimer
L'approximation
de
Eiorn
oppenheimer appelee
aussi
approximatiernadiabatique
est
la
premiere
,cesapproximations utilisdes
pour
la
rdsolution
deI'dquation de
Schnidinger,
dufait
que les noyaux sont plus lourdsque les 6lectr'ns,
les noyaux se ddplacent donc
t'ds
lentement par rapport aux 6lecfions.on
commencepar ndgliger le mouvement des.noyarx par rapport d celui des dlectrons,
ils
s,adaptentinstantandment aux ddplacements des premiers. Les noyaux apparaissent donc conmre
immobiles aux
yeux
des dlectrons.[4].
Cette approximationconduit d
une fonction
d'onde
6lectronique
qui
il,'
ddpend
des
coordonndes
des noya'x
que
parametriquement.
Leur
comportementn'est
pasr-
'
.*:
Chapitre
I
_
.fh6orie
de la
fonctionnelle
de la densit6(D
fh6orie
de ra fonctionnelre de ra densit6fDFr;
t(F
ddplacement.
Donc,
nous
ltouvons6liminer
le
terme
TN,ot
nous considdrorrs queUxn est constante. Ce qui permet de metke : [2]
He=Te*Uee*Uen
on
a donc rdduit la complexitd du probldme. Mais la solution de l,dquation deSchr<idinger reste toujours dilificile.
Il
faut faire d'autres approximationspour
r6soudre ce probldme.
[3]
L4.
L'approximation
deHartree
-
Fock
(rr.4)
Cette
approximation consiste
d
supposerque
chaque
electron se
diplace
inddpendamment dans un champmoyen
er66 par les autres dlectronset noyau<.
on
ramdne donc
le
problemerelirtif
dun
grand nombre d'dlectronsd un probleme d un
seul dlectron'
L'Hamiltonien
peut
Otreecrit
commeune
sornme des Hamiltoni,ens ddcrivant un seuldlectron. [4,1i]H:\iHt
On
lri:#
ai
+
ui(:ri) +
vt(rt)
Tel que
Ui(.i=
-Xu,#
L'dnergie potentielle de l'dlectron
i
dans les champs de tousres noyaux k.
u(ii)
-
L/zEi#
C'est le champ
effectif
de Harb:ee.(r 5)
(r 6)
(r.7)
(r.8)
,f,fu
Chapitre
I
'rh6orie
dela
fonctionneuede ta densite
(DFT)
laqE:
'l'
Le
potentieleffectif
est la somme de cesdeux contributions
Y"fr@-vhf)+vn(i)
V
h
Le potentiel de Harhee.vn
Le potentiel d'interactio,n dlectron- touts aufies noyaux. En introduisant le potentielelfectif
dans l'dquation de scbnidinger.on
trouve-u2v2
v,(i)
+ v"tr?1y,(i)
_
sivi(i)
Ge)
La
fonction d'onde du
systdrme dlectroniquea
la
forme d'un produit de
forrclion d'ondes des dlectrons, et l'6nLergie de ce sysGme 6galed
la
somme des 6nergies de tous les dlectrons.
V (F
L,F2... .....,/n) =Y1 (f
1)v
Z(iz)
.......yn
(in)
E:E'+E, ..
...8n
L'dquation
(I'10)
estbien une solution de l'6quation
(I.9)
mais ne
respecte pas
le
principe
DePaal|
L'approxinution
de <Fliarhee-Fock
>
a 6t6 introduite pour prernclreen compte
Le
spin des 6lectrons pourla
rdsolution del'6quation
de schnidinger:..La
difference
entreL'dnergre
du
systdme multidlectroniquer€el,
et
l,dnergie
obtrenue
dans I'approximation
de
Harfee
cofirme 6tant
celle
reprdsentantle
reste
des interactions dlectroniques.L'une
de ces interactionsqui
manque dansle
moddlrs r1eHartree est I'echange et
la
corrdlation. L'dchange estd'origine
purement quanti,que.c'est
cet effet
qui
exprime
l'antisymdtrie
de
la
Fonction d,onde
par
rapport
dl'dchange des coordonndes
de
n'importe
quelsdeux
dlectrons menantd
ddcrir,e lesystdme d
N
corps (6lectrons) piarl'6galit6
:v(ri,ia,ib
......in)
=
y?L,ib,ia
...in)
Y
Doit
6tre
antisymdtrique. D)onc,elle s'dcrit
sousla
forme
d,un
ddterminant deSlater (r.10) (r.1 1)
*tlttJ
Yn(in)J
Y(rL,i
2...,i
n)
-+
[
*
t
!tt'
'
'tM'[v
nirrl
(r.12)
t-L5. Th6orie
dela fonctionnelle
dela
densit6(DFT)
Le
concept fondamentat de la fonctionnelle dela
densite est que l,6nergi,e d,unsysteme dlectronique peut €tre exprim6e en
fonction
de sa densite.
c,est
en farit uneid6e
ancienne datant principalement destravaux
de
Thomas
[6]
et de Fermi
[7].L'utilisation
de la densite dlectronique corrme variable fondamentalepour ddcrire les
propridtds
du
systdme
existe depuis
les
premidres approches
de
la
stnrchreelectronique de la matiere mais elle
n'a
obtenu de preuve que par la d6monstrationdes
deux th6ordmes dits de Hoherrberg et
Kohn.
[9]L5.l.Les
th6orimes
deEohenberg
etKohn
Le ddveloppement de la thdorie de la fonctionnelle
de la densitd a commencr5
dans les anndes 1964 et 1965 avec les publications de
Hohenberg
etKohntgl
(196,4).Les deux thdordmes sont comrne suit :
Th6orime
01L'dnergie totale
de l'etat
fondamentalE
estune fonctionnelle unique
<Ie
la
densitd des particules
p
(i)
pour un
potentiel externey"*,(i)
donn6e.
ce
thdorrernesignifie
qu'il
suffit
de
connaitre seulementla
densitd dlecffoniquepour d6termi'er
toutes les fonctions d,onde.
Th6orime
02La valeur minimale de lar fonctionnelle de I'dnergie totale
est l,6nergie de
l,e1't
fondamental
du
systdme.Ils
ont
montrd que
la
densitd
qui
donne cette
valeur minimale estla
densitd exacte del'dtat
fondamental d'une particule, et que lesaurtn:s
propridtds
de
l'6tat
fondamental
sont aussi
fonctionnelles
de
cette
densit6.
tg]L'dnergie
del'6tat
fondamentald'un
systdme dlectronique dans un potentiel ext6de*r
est ddterminde par la m6thode variationnelle.
l2j
7 'Th6orie de lafonctionnelle
dela
densit6 E[p0]=14inE[p0]
(r.13)'fh6orie
de lafonctionnelle
de
la
densitdL5.2.Les dquations de
Kohn-
ShamCes
dquations
ont
pour
objectif
la
ddtermination
des fonctions
d,ondesdlectroniques
?f
qui
mini;misent
l,6nergie totale.
Les
fonctions d,ondes
sont ddtermindesd
partir d'une
dquation
similaire
d
l'dquation de
schnidinger
d,unemanidre auto-coh6rente. L'dquation est donnde par
[10]
:l#vz
+vion@
+
vHG)
*vxc(i)l
*a
=
eiyi@
(r
14)
Y1(r): la fonction d'onde de l',6lesfien
i
V
(r)
*":
reprdsente le potentirll ionique.V,(r)
: repr6sente le terme de HARTREEdonndpar:
vHffl=
yeuet:t ditdiz
" trL-r2l
(L 15)Le potentiel d'6change-corrdlation est obtenu
dparttde
la d6riv6e de l,dnergie
d'dchange corrdlation par
npport dla
densite :E*
Vxcffl=
asxc[P(il]ap?)
(I. tr5)
Donc les 6quations de
KoHN-sHAMpeuvent
s,dcrire sous la forme:
HviO:[-
*o'
+
vetr(F)]v'i(D
-
eiyi(i)
(11,7)
ori
chaquedlecfon
subit I'effet; du potentieleffectif
crd6 par tous les noyaux et les
autres 6lectrons, ce potentiel esrtdonnd par :
v"ff A:vexr(fl+
f
#np7ilafi
+
vxcfi
Les orbitales des
K-s
sont ddcrites par l'expression suivante:
Yi6E4=2 jcij
q(i,n
(r.18)
(r.1e)
OJ
t[,a
sont les fonctions de berse.Cij
: sont les fonctions de base.Les solutions des 6quations K-S reviennent d ddterminer les coefficients pour les
orbitales occupdes qui minimisernt l'dnergie totale. Si les bases sont donn6es.
la
chapirre
r
r
p
matrice Hamiltonienne
H
et
dechevauchement,s sont construites, L,dquation
sdculaire est ddfinie comme suit :
(H-A€|S)C|=A
g.2:0)
Jusqu'ici
laDFT
est une mdthode exacte, mais pourque la
DFT
et les 6quationsde
Kohn-sham
deviement urtilisables dansla
pratique, on a besoin de proposer rrne
formule pour
E
"[p(r)]
et pour cela, on est obtigd de passer par des approximations.L6.Approximation
dela densit6locale
(LDA)
Dans
l'approximation
de la densitdlocale (LocatDensity Approximation LD,4),
Pour approximer
la
fonctionnelle dela
densitdE*" [p(r)J,
Kohn
et
Shamproposaientd6s
1965I'approximation
de
la
densit6locale
(LDA)
[11], qui traite un
systdrneinhomogdne comme dtant localement homogdne,
avec une 6nergie d,dchange et,ce corrdlation connue exactement
:
Exc'Dr
Jpli;
F
I
nb)r*cho*lt
(i)ldr3
(r.21)
of
exch*1p
7i11
designe I'dnergie d'6change-correlationpo,r
une particu,ted'vrr gazuniforme
d'dlectrons de densit6p
.v *
rtT
)
=
!ry#g
excho*
1p(fi1+,
1r1 a"."yLernt
(r.22)
Dans
le
cas des materiaux nnagndtiques,le
spin dlectroniquefournit
un
degrd delibertd
suppldmentaireet la
LDA
4oit
alors
etoe dtendue
d l,Approximation
de
ladensitd
de spin
Locale
(LSDA
:
Local spin
Density Approximation)
ori
l,6nergie:
d'echange
et
de corr6lationQ"
devient unefonctionnelle
des deux densitds despinL
hautetuas
p
(t)
.t
p
(J)
ExctsD,[pl,pJ]:
!
exc(pt
tflp
I
(r)pl)ar,
9
Chapitre
l
:
F
gu t"LSDA@
T?)p
J est l'rSnergied'echange-condlation par
particule d,un saz
d'dlectrons homogdne.
L7.La
m6thode desgradients
g6n6ralis6s(GGA)
Malgrd la
simplicit6
de laLDA,
elle a donnddes rdsultats fiables dans plusieurs
cas' mais
ils y
avaient des cas orielle dtait
en contradiction avec l,expdrience.
pour
cette raison
le
gradient
de
la
densitd
d'dlectron
a
eteintroduit
conduisant
dI'approximation du
gradient E;endralis6GGA ori
l,6nergieE*"
est enfonction de la
densitd d'dlectron et
de
son gradientVo6
ExcGGAfp(Dl:
I
p(fl
eoor,,xc (p
(r)V
p@
dr3
(r.24)
La
GGA
est donn6e par diffdrentes paramdtrisations,parmi elles celles de perdew et
ces collaborateurs [12,131.
L8.Solution
del'dquation
de tr(ohnet Sham
Aprds
la
ddtermination
d*
terme
d'dchangeet
de
corrdlation,
il
nous
restemaintenant de rdsoudre I'dquatirm de
Kohn
etSham:
H"o@i(r)
:
ei<Di(r)(r.25)
Tel quetH"p=L-#o:
*
ftr ff
or'
*
vxc
+
vextlet'hamiltonien
dekohn
et
Sham
Pour une seul particule (single particle).L-#rr
*
*t
ffior'
*
v:uc+
vextlai?)
=
eicDi(r)
(r.26)
Les
mdthodes basdes surla DFT
sontclassdes suivant les reprdsentations qui
sont utilisdes pour la densit6, le potentiel et les orbitales
de
Kohn
etsham.plusieurs
choix de
la
reprdsentation sontlbits
pour minimiser
le
cott
decalcul
en termes detemps en maintenant suffisammerrt la prdcision.
Chapitre
.
-
I
-
-th6orie de ra fonctionneuede Ia
densit6,rrD
CF
L9. L'auto-coh6rence
dans tles calculsPour
simplifier
les calcrils,En
r6solvant les 6quations deKS pour
les points de
sym6trie
dansla
premiere zone
de Brouillon.
ces
solutions s,obtiendront d,ure
manidre iterative
en
utilisant
un
cycle
d'iterations
auto-cohdrent
illustre
par I'organigramme de laI.Fig.
1.on
commence par une densitdd'essai
pi,
wur
la premidre
itnration.Typiquern.ent onutilise
une supe{position dr:s densit6s atomiquespuis on calcul la matrice de
Kohn
sham' et en rdsolvant les 6quirtions pour lescoefficients d'expansion pour obtenir les
orbitales de
Kohn-sham,
d
cette dtape,en
calcul antla
nouvelle densit6
p.*. si
la
densitd
ou
1'6nergiea
beaucoup chang6(criGre de
convergence),
on
retourned
lapremier
6tap,
et en
mdlangeantles deux
densitds
de charge
pin
et
g*t
de
la manidre suivante : [14]Pli'=(r-o)pl"*opl,,t
(r.2i')
i
reprdsente laie.itdration
a
un paramdtre de mixage.Ainsi la
procddure itdrative peut €tre poursuiviejusqu'd
ce que
la
convergence soritrdalisee'
on
peut reprdsenter cette procddure par leschdma ci-apres.
r--T'
I
'l
il?-i;1
"t""*t-"
cycle
auto cohdrent de la thdorie de la fonctionnellede densitd
lfh6orie
de lafonctionnelle
de Ia densit6,#L
chapitre
r
Th6orie
de lafonctionnelte
de ta densit6
(DFT) Gt
Bibliographie
[l]
E'schrodinger an undulatory of the mechanics ofatomes and moldcules phys.rev
.v o1.28,1926,p,1 0 49
-l
A7 0[2]
Ivldmoire de magister universitd dem,sila
pr
kouriche athmane,2006
[3]
M.Born,
J.R. Oppenheimer,l\nn. phys. g7,457 (1927).[4] M6moire universit6 de
m'sila
,de magister parmeuagfalita
,2A03
[5]
s.J. Lee, T.s.Kwon, K.
Nahm.,c.K. Kim,
J. phys. condens. Matter 2
(rgg0)3253.
[6]
M6moire de magistdre, universitd Mohamed Boudiafde m,sila,
Baraka
Fatiha,200,4[6]
L.H.Thomas, pro. Combridgephilos.
Sloc.23,542 (1927).[7] E. Fermi, Z.
phys.
48,73 (192St).[8] P. Hohenberg, W. Kohn" phys. Rev.
B
136,g64 (1964).
[9]
Mdmoire
universite dem'sila
de magister
parzerargafares,200g
[10] W. Kohn, L. J. Sham, phys. Rev.
A
1t33,140
(1965). [11] L.J.Sham, W.Kohn, phys.Rev t4S(tg66)
561u2lJ'
P' Perdew, J.A
chwory,
s.H. vosko, K. A.
Jackson,
M. A.
perderson, D. J. Siqghand C. Fiolhais, phys Rev.
8.46'O992)
6671.[3]
J. P.Perdeq
s. Burke andM.
JErnzerhof,phys.
Rev.Let.77 (1996) 3g65.[14]
s'
conenier, Densiry FunctionaflTT:w
and the
family of (L) Apw-methods:
a
Chanitre
.--- M6thodesd
II
p
Ir'
Mcthodes
drl
calcul
des
structures
de bandes
6lectroniques
fntroduction
Pour
comprendreles
differentes
propri.tes des
solides,
il
estd'dtudier
le
comportementdes dlectrons dans ces derniers,
et
ceci est enavec les m6thodes de calcul qu,on classe en trois groupes :
n6cessaire
lien
directLes
m6thodes empiriques
pour
lesquelles
les
calculs
ndcessitentdes
resultatsexpdrimentau:<' semi-empirique
pour
lesquellesles
calculs ndcessitantd la fois
desrdsultats experimentaux et deti donndes fondamentales.
Et enfin
celles ditesab-inifio
pour lesquelles les calculs n6cessitent seulement les donn6es
fondamentales.
Les
techniquesde calcul de
la
structure 6lectronique mises
au
point
au
cours 6esdernidres d6cennies sont nombreuses, et en particulier, les mdthodes
ab-initioqui
s'nt
devenues
aujourd'hui un
outil
de basepour
le
calcul
des propridtes dlectroniques etstructurales des systdmes les p,tus complexes. Elles
sont aussi un
outil
de choixpou'
laprddiction de nouveaux mat6niaux, et
elles ont parfois
pu
remplacer des exp6rienc;estrds cotteuses ou m6me irrealirnbles en laboratoire.
Les
dtudes
ab-initio
rnendessur
l'ensemble
des
matdriaux existants
sontnombreuses'
et ont
donn6 des r6sultatsfiables
en
le
comparantavec les
mesures expdrimentales.parmi
ces
mdthodesab-initio
[rJ la
mdthode des ondes
planssaugmentdes lindarisees
(Fp-LApw)
estl'une
des plus pr6cises, actuellement,pour le
calcul de
la
strucfure dlectrorriquedes solides
dansle
cadrede
la
th6orie
de
[afonctionnelle de la densit6 (DFT).
Elle
est semblable d la mdthode
Apw
avec tous lesavantages de la m6thode
oPW
pour traiter les semi-conducteurs.Ainsi, la
rapidit6dle
calcul de la m6thode
FP-LAP\['est
impressionnante par rapport aux autresmethoders
de premier principe.
Dans ce chapitre,
on
exr)ose d'unemanidre
g6n6rale les differentes mdthodes de calcul des strucfures de bandes.-"1
Chapitren
Mfthodesd
p
Itl.
Mdthodes decalcul
dld:mentaires[I.1.
M6thode
decalcul
des 6lectronslibres
ce
moddrefut
l'eurre
parSommerferdtzlqui
constitue re point de ddpart de
la Physique
del'etatsolide.
F'our cemoddle, le potentiel d,interaction, dans l,equation
de
schr<tdinger, estnul' car
cet dlectron ne pergoit aucune influence de
la
part
des
autres dlectrons environnants.
on
peut dire
que
lorsquel'6lectron
est
libre,
tous les niveaux
d,dnergie
lui
srontpermis. Ces niveaux forment une bande continue.
Dans
ce
moddle,
les
6lectronsles moins li6s aux
atomes
du m6tal
se
ddplacent librement danstout le volume'
Dans cette approximation, on ndglige les
interactions
entre
les
6lectronsde
condu'ctionet les
cations
:
tous
les
calculs sont
faits
avecI'hypothdse
of
les
dlectronstle
conduction sont
libres de
se ddplacern,importe
ot
dans
le
cristal- L'.nergie totale
ne
comprend
que l'dnergie
cindtique,
l,energiepotentielle
est
n6glig6e.
L'dtude
du
moddre
de
sommerfeld
t3l
permet
d,dcrire l'dquation de Schrddinger d,urLe particulesous la forme :
#(#*#*f;*,0,
Le Moddle des dlectrons libres est cependant incapable d'expliquer pourquoi
certains
solides sont
des semi-conduclnursou
des isolants.c,est
l,un
de
sesplus
granas d6fauts.ILl.z.
M6thode
des 6lectrons presquelibres
CN.F.E.N4
ce
Moddle donne de bons rdsultats, pourles mdtaux dont les
ceurs
ioniques nesont pas en contacf
[4]'
Pour les m6taux noblesdont les c@urs se touchent, un
autre
moddle est ndcessaire.
Dans
le
moddle, des dlectrons presque libres, lesdlectrons de valence
qui
forment 1:gaz D'electrons
de
conductiorLvont donc
baigner dans
un
potentiel
periodiquecristallin t'ds faible'
Dans
ce
c)€ts,on
considdrele
champ periodiquecorlme
'n*
perturbation.
(r1)
Chanitre
ll
M6thodesd
r
Ct""t.ooiqu.,
(ffi
Dans la mdthode des 6lectrons presque libre, l'dnergie d,intemr;tion
de l,dlectron iavec
le
champperiodique
du
rdsreaucristallin
est
consid6r6e beaucoupprus
faible
quel'dnergie
cindtique deI'dlectron
et que
I'influence
du potentiel cristalrinest anar.gue
a une perturbation periodique
H:E+V14
Avec
v(r):w
(r)
: perturbationtr
2. M6thodes decalcul
avanc6esII.2.I
La
m6thode des liaiisonsfortes
(L.C.A.O)
La
mdthodeL'c'A'o
(Linear combination
of Atomic
orbj.tals) est basee sur la mdthode desliaisons fortes' I)ans
cette approximation,re
potentier resteun
tefineimportant
devantl'dnergie
ci'dtique
des 6lectrons
et
peut €tre d6crit comme
une
sornme de potentiels atomiques.
[4]
cette
mdthode donne
de
bons rdsultats lorsque
les
orbi&rles atomiques so.ntlocalisdes autour des noyaux et changent peu lorsque
res mornes sont rapproches pour
constifuer te cristal'
ceci
estwai
pour les 6tats duceur
de l,atome mais l,est beaucoupmoins pour les dlectrons de valence
En
outre, en cequi
concerne les 6tats de valencela
mdthode estmieux
adaptdeau calcul des 6tats assocides aux combinaisons liantes,
c'est-d-dire
d
la
bande de valence, qu'auxdtats rdsultant des
combinaisons
liantes ,
c'est-d-dire d la bande de conduction.[5]
L'hamiltonien
total du cristal perrt €tre approximed
I'hamiltonien
dte l,atome isol6.H=Ha=#o*voe)
(r
3)
va(r)
: I'dnergie potentielle de l'd,lectronli6
d l'atome consid6r6.
En rdsum6' cette mdthode est
bien
adapteeau calculdes bandes profondes dtroites,
rdsultant de I'dlargissement des 6tats
du c@ur, un peu
moins
adapt6eau calcul dela
bande de valence et peu recommandde au calcul
de ra bande de conduction.
(rr.2"l
chapitre
tr
Mdthodes drlcalcul
desstructures
q.
b".d*s!q.,.ooiou",
GF
tr
3.M6thodes des ondes planes:ceue
mdthode constitue la base d'un ensemble de m6thodes dites des ondes planestelles que
Ia
mdthode des o.ndes planesorthogonalisdes
et celle
des ondes planesaugmentdes.
Pour rdsoudre I'dquation
de
schnidinger
v=
Ey
en tenant compte der la pdriodicitd du rdseau
cristallin,
on choisitune forme particulidre de la fonction d,onde
6tablie par Bloch.
Vr(r) =
Up(r)exp
(ik
r)
0r
4)Pour le calcul de la relation de dispersion Eo
(k)
onfait
appel d ldquation s6culaireoi
la connaissance de I'expression ddcrivant lepotentiel cristallin est n6cessaire.
D'aute
part, ce calcul exige que le nombre dbndes planes utilisdessoit suffisamment
grand ce qui rend la t6che diffi<;ile,
d'ot
on obtientune faible convergence. [6]
Il3.l.M6thode
desonde
planes orthogonalisdesLa
mdthodeLCAO
procede de I'idde que les6tats 6lectroniques dans
le
cristalsont
essentiellementdes
6tats atomiques,plus au moins
perturb6s
par
la
naturep6riodique du cristal.
La
mdthode
opw
(orthogonalized
prane
waves)
procdde
de
l,idde
diamdtralement
opposde'
que
les
6tats
6lectroniques
danrs
Ie
cristal
sontessentiellement les 6tats dans l'6lectron
libre, plus ou
moins perlurb6s
par
lanature p6riodiquedu cristal les
foncfironsd'onde des 6lectrons
6
dansIe
cristal
sont alorsd6veloppdes sur la base des fonctions d'ondes des 6lectrons
libre,
c,est d
dire
d,onde;splanes'
Il
en resulte quele
domidne devaliditd
de cette methode est compldmentairt:du domaine de
validitd
que celler deLCAO. cette
mdthodeest
adatfiked l,etude de Iabande de conduction,
un
peu moinsii
celle
dela
bande de valenrce et pas dutout
drcelles des 6tats du
caur.
La m6thode des ondes planes orthogonalis6es a dtd proposee par
Herri ng
en
1940[7]' Depuis
safonnulation'
elle est devenue une des methodes les plusutilis6es pour
Chapitre
ll
M6thodesdr
erc.trooiqu.,
ffF
le
calcul
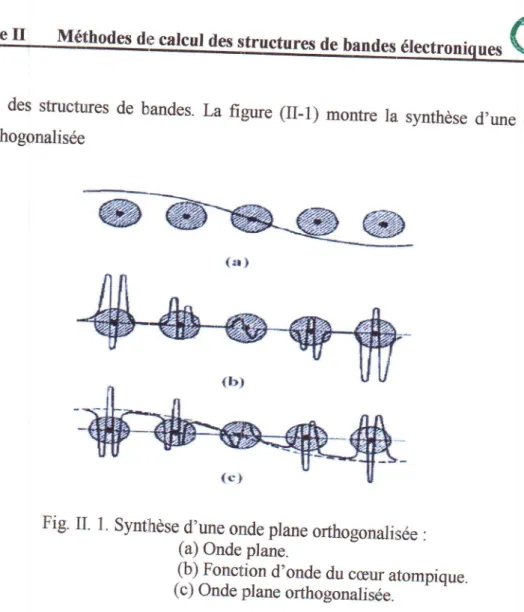
des structures de bandes. plane orthogonaliseeLa figure (II-1)
monfie
la. synthesed,une
ondeFig.
II.
I.
Synthesed.*
onde plane orthogonalisde:
(a) Onde plane.
(b) Fonction d'onde du
ceur
atompique.(c) Onde plane orthogonalisde.
IL3.2. La
m6thode des pseudopotentielsLa
mdthode pseudopotentieflecoilrme
ra
mdthodeopw
utilise
d'orthogonalit6 des dtats de valenceet
de conduction avecles
titats
I'effet de
I'orthogonalitd est
inclus
dansle
potentiel
sous
ra
formedquivalent appel6 pseudopotenti,el.
Philips Kleinman [9J a demontrei, avec son thdordme d,annulation, que les elechons drl
valence prdsentent expdrimentalement un potentiel
rdpulsif
lorsqu,ils sont proches duccur
ionique'ce
potentiel repulsif agit pour retenirles 6lectrons de valence en dehors
du
ccw'
of
en d'autres tennes, les fonctions d'ondes des dlecfions sont contraintesdr
6tre orthogonalisdes aux 6tats du
ceur.
Une
fois
le potentielrdpulsif
sonrmd avecle potentiel attractif des 6lectrons du
caur.
ceci engendre un potentiel trds