Adrenal cancer: ESMO Clinical Practice Guidelines for
diagnosis, treatment and follow-up
†A. Berruti
1, E. Baudin
2, H. Gelderblom
3, H. R. Haak
4, F. Porpiglia
5, M. Fassnacht
6&
G. Pentheroudakis
7on behalf of the ESMO Guidelines Working Group*
1
Medical Oncology, Department of Clinical and Biological Sciences, University of Torino, Azienda Ospedaliero, Universitaria San Luigi, Orbassano, Italy;2
Service de Médecine Nucléaire et de Cancerologie Endocrinienne, Institut Gustave-Roussy, Université Paris XI, Villejuif, France;3
Department of Clinical Oncology, Leiden University Medical Center, Leiden, The Netherlands;4
Department of Internal Medicine, Máxima Medisch Centrum, Eindhoven, The Netherlands;5
Urology, Department of Clinical and Biological Sciences, University of Torino, Azienda Ospedaliero, Universitaria San Luigi, Orbassano, Italy;6
Department of Internal Medicine I, Endocrine Unit, University Hospital, University of Würzburg, Germany;7
Department of Medical Oncology, Medical School, University of Ioannina, Greece
incidence and epidemiology
Two different primary malignancies can arise from the adrenal gland: the adrenocortical carcinoma (ACC) from the adrenal cortex and the malignant pheochromocytoma from the adrenal medulla. Both malignancies are extremely rare. ACC has an estimated incidence of∼0.5–2 new cases per million people per year. It follows a bimodal age distribution, with peaks in childhood and in the fourth tofifth decades of life. ACC is more frequent in women than in men (ratio 1.5∶1). The majority of ACCs are sporadic; however, sometimes these malignancies form part of hereditary syndromes such as the Li-Fraumeni syndrome, Beckwith-Wiedeman syndrome, multiple endocrine neoplasia (MEN) 1, congenital adrenal hyperplasia, familial polyposis coli, and B-catenin mutations [1]. Germline p53 mutations without Li-Fraumeni are frequent in southern Brazilian children.
The frequency of sporadic ACC cases is consistently greater in the population of patients with adrenal incidentaloma (2% in the most conservative series) and, in our experience, the proportion of incidentally discovered ACC is increasing.
Pheochromocytomas are catecholamine-producing neuroendocrine tumors arising from chromaffin cells of the adrenal medulla or extra-adrenal paraganglia. In the latter setting, the term paraganglioma is preferred. Their incidence is ∼2–8 per million adults per year [2]. The incidence of pheochromocytoma increases to 0.5% in patients with hypertensive symptoms and can be as high as 4% in patients with adrenal incidentalomas. Up to 30% of
pheochromocytomas are associated with a variety of inherited conditions, including MEN2, Von Hippel–Lindau (VHL) disease, neurofibromatosis type 1, and heredity paraganglioma syndromes. Only a few (10%–17%) pheochromocytomas are malignant. Although the likelihood of malignancy varies
among different genetic backgrounds, it is below 10% for most sporadic pheochromocytomas, except in patients with
mutations in the succinate dehydrogenase B (SDHB) gene and/ or extra-adrenal locations, among whom more than 30%–50% may develop a malignant tumor.
diagnosis and pathology/molecular
biology
Somatic mutations of the tumor suppressor gene TP53 are observed in one-third of ACCs. In addition, allelic losses (LOH) at the TP53 locus (17p13) are observed in >85% of ACCs. The insulin-like growth factor II (IGF-II) locus (11p15) is imprinted and IGF-II is overexpressed in 90% of ACCs. About one-third of ACCs harbor somatic activating mutations of the B-catenin gene [3].
The gene encoding subunit B of theSDHB complex is by far the most important contributor to a hereditary malignant pheochromocytoma/paraganglioma. In addition, many sporadic metastatic pheochromocytomas/paragangliomas have similar molecular profiles to those of hereditary tumors. Inactivation of mutations inSDHB is thought to reduce function of the SDHB complex, causing a pseudohypoxic state and increased expression of angiogenic, growth and mitogenic factors via stabilization of the hypoxia-inducible pathway. If no SDHB mutations are identified, genetic testing for VHL, SDHD, or succinate dehydrogenase C (SDHC) is indicated and may provide future insights into the best therapeutic options to be developed for these patients [2]. Rearranged during
transfection (RET) should be tested in case of calcitonine secretion and may be tested when all other gene mutations are negative.
A detailed preoperative endocrine assessment is essential to establish the origin of the tumor (cortex versus medulla versusothers). In all cases of an adrenal mass a comprehensive hormonal analysis is strongly recommended. In 2005, the ACC working group of the European Network for the Study of Adrenal Tumors (ENSAT;www.ensat.org) proposed standards for this situation in patients with suspected or established ACC
†Approved by the ESMO Guidelines Working Group: June 2012.
*Correspondence to: ESMO Guidelines Working Group, ESMO Head Office, Via L. Taddei 4, CH-6962 Viganello-Lugano, Switzerland. E-mail: clinicalguidelines@esmo. org
clinical
pr
a
ctice
id
li
© The Author 2012. Published by Oxford University Press on behalf of the European Society for Medical Oncology. All rights reserved. For permissions, please email: journals.permissions@oup.com.
(Table1). It is important to acknowledge that the evidence level for this proposition is formally low, although the diagnostic accuracy is high (V, B). Furthermore, the
preoperative hormone pattern may serve as afingerprint of the tumor during follow-up. Whereas the measurement of metanephrine and normetanephrine is well established for any pheochromocytoma, plasma and urine methoxytyramine levels that provide useful information to assess the likelihood of malignancy [4]. However, this marker is not yet widely available.
Urine steroid metabolomics revealed a pattern of nine predominantly immature, early-stage steroidogenesis in ACC that performed well in differentiating adenomas from ACCs. The use of this technique in clinics is, however, premature [5].
For best patient care, adequate visualization of the tumor and potential metastases is essential. For differential diagnosis of an adrenal mass computed tomography (CT) and magnetic resonance imaging (MRI) are currently considered equally effective. CT is less expensive and should be recommended as thefirst choice in a suspected ACC. Conversely, MRI offers a preferential application for the suspicion of
pheochromocytoma due to the risk of a hypertensive crisis after infusion of i.v. contrast medium for CT. Although these methods cannot determine the exact entity of the mass, both are able to correctly diagnose benign tumors in most cases— when performed according to the state-of-the-art criteria (e.g. Hounsfield units <10 in unenhanced CT, rapid washout in 15-min delayed contrast-enhanced CT or signal intensity loss using opposed-phase MRI strongly suggests a benign tumor). Most ACCs and malignant pheochromocytomas are
inhomogeneous with irregular margins and irregular enhancement of solid components after the infusion of i.v. contrast medium and, in many cases, it is difficult to distinguish these tumor entities using conventional imaging. The detection of local invasion or tumor extension into the inferior vena cava, as well as lymph node or other
metastases (lung and liver), is important for planning surgery. F-18/flourodeoxyglucose-positron emission tomography (FDG-PET) is a useful tool for distinguishing potentially malignant lesions from benign tumors in radiologically indeterminate adrenal lesions. In patients with established pheochromocytoma, FDG-PET appears to be superior to metaiodobenzylguanidine (MIBG) and
somatostatin- or F-dopamin-based methods, particularly in patients with SDHB mutation and malignant tumors, and should be indicated as the preferred method [6] (IV).
Fine needle biopsy of a suspected ACC is almost never justified because of anticipated tumor spill, and in suspected pheochromocytoma, it is contraindicated.
The pathological differential diagnosis of adrenal neoplasias is still largely based on morphological features requiring an experienced pathologist. Several markers have been introduced to establish the adrenocortical origin of adrenal masses, with steroidogenesis factor-1 immunohistochemistry being particularly useful [7], although not yet widely available. The differential diagnosis between carcinoma and adenoma is challenging as no single marker indicates malignancy. The most widely used diagnostic score has been introduced by Weiss et al. [8] and includes the following parameters: mitosis, atypical mitosis, necrosis, venous invasion, sinusal invasion, capsular invasion, nuclear atypia, diffuse architecture, and clear cell. A score of≥3 suggests malignancy. Ki67 as a marker of proliferative activity can also be useful in this respect.
For pheochromocytomas, the situation is similarly demanding. Several histologic features (local invasiveness, growth pattern, presence of necrosis, cellularity, spindled morphology, nuclear pleomorphism and hyperchromasia, mitotic activity, and atypical mitosis) comprise the Pheochromocytoma Adrenal gland Scaled Score. However, there is currently no consensus on the adoption of a formal scoring system for these tumors [2]. According to the current World Health Organization (WHO) classification, malignancy is defined by the presence of metastases to a site where pheochromocytoma/paraganglionic tissue is not normally present, e.g. liver or bone, to avoid confusion with multiple primary tumors. No single histologicalfinding can predict metastatic disease. However, patients with large tumor size (>5 cm), extra-adrenal location, or SDHB mutation have a higher risk of malignancy.
Table 1. Diagnostic work-up for adrenal cancer Hormonal work-up
Glucocorticoid excess (minimum 3 of 4 tests) Dexamethasone suppression test (1 mg, 23:00 h) Excretion of free urinary cortisol (24 h urine) Basal cortisol (serum)
Basal ACTH (plasma)
Sexual steroids and steroid precursors DHEA-S (serum)
17-OH-progesterone (serum) Androstenedione (serum) Testosterone (serum)
17-beta-estradiol (serum, only in men and postmenopausal women) 24-h urine steroid metabolite examination
Mineralocorticoid excess Potassium (serum)
Aldosterone/renin ratio (only in patients with arterial hypertension and/or hypokalemia)
Catecholamine excess
Normetanephrine, metanephrine, and methoxytyramine (plasma) Alternatively: fractionated metanephrine excretion (24 h urine) Imaging
CT or MRI of abdomen and CT thorax
Bone scintigraphy (when suspecting skeletal metastases) FDG-PET (optional)
MIBG scintigraphy, DOTA-TATE-PET, Dopa/Dopamine PET or FDG-PET if pheochromocytoma is proved
Adapted according to the recommendation of the ACC working group of the European Network for the Study of Adrenal Tumors (www.ensat.org/ acc.htm), May 2005.
In patients with a clearly established diagnosis of an ACC, one can skip the workup on catecholamine excess (and conversely for established pheochromocytoma, one can skip the steroid analysis). DHEA, dehydroepiandrosterone.
staging and risk assessment
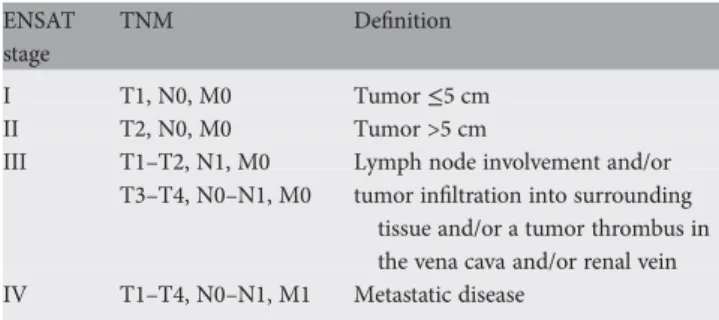
The disease stage and margin-free resection are the most important prognostic factors in ACC. In the assessment of the disease stage, we recommend the tumor–node–metastasis (TNM) classification proposed by the ENSAT network (Table2) [9] (IV, A), since this system seems to be superior to the staging system published by the Union Internationale Contre le Cancer in 2004. Age, mitotic count, the proliferation marker Ki67, and the glucocorticoid excess are additional prognostic parameters to refine the prognostic stratification [10]. New molecular markers for aggressiveness and survival have been recently proposed, but their use in clinics needs validation [11]. There is no staging system for malignant pheochromocytoma and, in contrast to other neuroendocrine tumors, no poorly differentiated category is recognized. The survival rate depends mainly on the tumor size, primary tumor location (extra-adrenal location is associated with poor prognosis) [12], and the SDHB mutation status (SDHB mutations are predictive of poor prognosis) [13]. The overall 5-year survival rate is between 34% and 60%; however,
heterogeneous survival is a critical characteristic of this tumor [12]. A significant number of patients die of hormone-related complications (increased blood pressure, constipation) [12].
management of local regional disease
Surgery is of utmost importance in the treatment of both ACC and pheochromocytoma. Adrenal surgery should be performed only in selected centers with >10 adrenalectomies for adrenal cancer per year [14] (IV, A).Open surgery with transperitoneal access is the standard treatment of all patients with localized (stage I–II) and local advanced stage (stage III) ACCs when complete resection can be achieved. Laparoscopic adrenalectomy is a safe and effective procedure for pheochromocytoma and a selected group of patients with small ACCs (<8 cm) without preoperative evidence for invasiveness and adrenal masses (e.g.
incidentalomas) that are judged as only potentially malignant. This technique must be performed only in centers with a consolidated experience in laparoscopic adrenal surgery, in which principles of oncologic surgical treatment are strictly respected [15] (IV, B).
The resection status (R0, R1, R2) is a major predictor of prognosis for ACC. A margin-free complete resection (R0 resection), in fact, provides the only means to achieve
long-term survival [16] (V, A). To obtain an R0 resection of a locally advanced ACC, it is often mandatory to resect ( parts of ) adjacent organs such as the wall of the vena cava, liver, spleen, colon, pancreas and/or stomach (V, B). Locoregional lymphadenectomy improves tumor staging and seems to lead to a favorable oncologic outcome [17] (IV, A). No data on the prognostic role of radical surgery and lymphadenectomy are available up to now for malignant pheochromocytoma.
For ACC tumors not invading the kidney, concomitant nephrectomy seems not to improve disease-free and overall survival and can be avoided, although additional data are needed (V, B).
In the case of inoperable local infiltrating or metastatic ACC, surgical excision of the primary tumor and/or metastasis should be considered in case of an objective response after neoadjuvant chemotherapy, when a radical resection seems to be feasible [16].
Cytoreductive (R2) resection in malignant
pheochromocytoma may improve the quality of life and survival by reducing the tumor burden and controlling hormonal hypersecretion [2] (V, B). It is also indicated in selected ACC patients with advanced tumors and severe symptomatic hormone excess when medical therapy is not able to control endocrine symptoms, and life expectancy is >6 months [18] (V, B).
Surgery for the recurrence of ACC (local and/or with a low tumor metastatic burden) after primary treatment might be effective in improving survival, if R0 resection is achievable and the time tofirst recurrence was >12 months (IV, B).
The hormone hypersecretion can increase the risk of perioperative complications. For ACC patients, it is very important to diagnose any adrenocorticotropic hormone (ACTH)-independent glucocorticoid excess prior to surgery to adopt measures to prevent a postoperative adrenal crisis or insufficiency. In all patients with glucocorticoid excess hydrocortisone must be administered during surgery (e.g. 150 mg/days) and postoperatively.
In patients with pheochromocytoma and secreting paraganglioma, exposure to high levels of circulating
catecholamines during surgery could cause hypertensive crises and arrhythmias. Therefore, all patients with
pheochromocytoma or paraganglioma should receive
preoperative preparation at least 10–14 days before surgery (V, A) [19]. Blood pressure targets for the treatment are <130/80 mmHg in the supine position, and systolic blood pressure preferably >90 mmHg in the upright position. Traditionally, the non-competitiveα-adrenoreceptor antagonist
phenoxybenzamine is frequently used. A standard dose is 10 mg twice daily with adjustments every 2–4 days. Alternatively doxazosine, a competitive and selectiveα1-adrenoreceptor antagonist might be as effective with fewer side effects. If the target blood pressure is not reached, Ca-antagonists (nifedipine slow release) or metyrosine may be used. Blockade of β-adrenergic receptors is indicated in patients developing tachyarrhythmias, but should never be started before the blockade ofα-receptors.
Hypertension during surgery may be treated with magnesium sulfate, intravenousα-adrenoreceptor antagonist
(phentolamine), calcium antagonist, and/or nitroprusside or
Table 2. Disease staging system for adrenocortical carcinoma ENSAT
stage
TNM Definition
I T1, N0, M0 Tumor≤5 cm
II T2, N0, M0 Tumor >5 cm
III T1–T2, N1, M0 Lymph node involvement and/or T3–T4, N0–N1, M0 tumor infiltration into surrounding
tissue and/or a tumor thrombus in the vena cava and/or renal vein IV T1–T4, N0–N1, M1 Metastatic disease
nitroglycerine. Tachycardia can be treated with intravenous β-adrenergic receptor blocker (esmolol). Postoperative hypotension should be prevented by saline infusion the day before surgery. If this adverse event occurs, it should be treated aggressively.
Postoperative care should also focus on glucose levels because hypoglycemia may occur after the reduction of catecholamine levels.
Most ACC patients have resectable disease at presentation, however, more than half of the patients who have undergone complete removal of the tumor are destined to have a relapse, often with metastases [1]. Similarly, radical resection is not a guarantee of cure for pheochromocytomas either. In one series of 171 patients followed up after surgical resection of a chromaffin cell tumor, 29 patients (17%) had recurrent or new tumors, which were malignant in 15 (9%) cases [20].
The aggressive behavior and the high recurrence rate of ACC provide the rationale for the use of adjuvant therapy. Adjuvant radiotherapy to the tumor bed should be considered in patients with incomplete/R1 resection or Rx resection [21] (V, A). Mitotane has been the reference drug for the management of ACC for decades. In a case control study involving 177 patients, the outcome of 47 patients followed in Italian reference centers that systematically adopted adjuvant mitotane to all radically operated ACC was significantly higher (in terms of both disease-free survival and overall survival) than the outcome of 55 Italian patients and 75 German patients followed in institutions not administering adjuvant mitotane therapy [22].
Although these data cannot be considered conclusive, they suggest that adjuvant mitotane can delay and possibly prevent a recurrence of disease (V, B). On these bases, a panel of international experts unanimously stated that patients with potential residual disease (R1 or Rx resection) and/or Ki67 more than 10% should be offered adjuvant mitotane, whereas adjuvant therapy was not considered mandatory in patients fulfilling all of the following criteria: stage I or II disease, histologically proven R0 resection; and Ki67 expressed in ≤10% of neoplastic cells [23] (V, B) (Figure1A). For these patients participation in thefirst prospective multinational randomized trial, testing the efficacy of adjuvant mitotane therapy (www.adiuvo-trial.org) is recommended whenever possible. Mitotane is a difficult drug to manage, with a long half-life, dose-limiting toxicity, and a narrow therapeutic window. Although the evidence is limited, the panel recommends that the mitotane dosing should be guided by plasma measurements, aiming at a concentration between 14 and 20 mg/ml since there is less antitumor activity <14 mg/ml and the risk of toxicity (gastrointestinal and neurologic) is higher above 20 mg/ml (V, A) [24]. A suggested mitotane regimen is depicted in Table3. Several blood analytes should be monitored during mitotane therapy. Due to the adrenolytic activity of mitotane, all patients must receive concomitant administration of glucocorticoids to cover adrenal insufficiency. Owing to an increased metabolic clearance rate of
glucocorticoids by mitotane therapy, high-dose glucocorticoid replacement is needed. Mineralocorticoid supplementation is necessary only in a subset of patients (Table3). Testosterone and thyroxine supplementation may be also required. Mitotane can also induce a wide spectrum of hormone and metabolic derangement and so an experienced endocrinologist should be
involved in the follow-up of patients during mitotane treatment.
There are no data regarding the optimal duration of adjuvant mitotane; we recommend that adjuvant mitotane should be administered for at least 2 years since the greatest frequency of disease recurrence is expected within this timeframe. It is noteworthy that while mitotane is well tolerated by a fraction of patients, the majority of patientsfind mitotane a difficult therapy that markedly impacts the quality of their lives. So the treatment duration should be assessed individually, taking carefully into account the cost/benefit ratio.
management of advanced/metastatic
disease
Prognosis in locally advanced inoperable and metastatic ACC patients is poor, the 5-year overall survival being <15%. However, recent studies have highlighted a greater heterogeneity of advanced ACC than previously thought. Extremely long survival has been reported in patients with resectable oligometastatic disease with considerable intervals among recurrences [10] (V, B).
Figure 1 Algorithm on management according to stage, risk factors, and disease characteristics for adrenocortical carcinoma (ACC) (A) and pheochromocytoma (B). *Low-risk ACC is defined stage I–II and Ki67 expression in≤10% of neoplastic cells, high-risk ACC: stage III or Ki67 expression in >10% of neoplastic cells.
Mitotane is the only drug approved in locally advanced inoperable and metastatic patients although randomized, controlled prospective trials are lacking (IV, A). Response rates vary between 13% and 35%, but many of these results were derived from retrospective series since the 1960s with variability in the response criteria [1, 10]. Patients with long-term maintenance of mitotane levels in the therapeutic range may obtain a survival benefit [25]. Patients with hormonal excess often experience clinical benefit of this strategy and continuation of mitotane treatment can be indicated in these patients even after radiological progression when alternative strategies to inhibit the hormonal excess are lacking.
Owing to the latency of mitotane to attain the therapeutic range, mitotane monotherapy is indicated in the management
of patients with a low tumor burden and/or more indolent disease (Figure1A). The combination with locoregional therapies such as radiofrequency ablation (RFA) is recommended. In case of rapidly progressing or life-threatening extensive metastatic disease and/or radiological progression under mitotane, cytotoxic chemotherapy is indicated. Over the last 15 years, only 11 prospective single-arm chemotherapy studies with a total of 239 patients with or without mitotane have been published. Response rates vary between 7% and 54%, again with variability in the response criteria. The association of mitotane to chemotherapy seems to be more active than chemotherapy alone [1], although no randomized trials have formally demonstrated this superiority. In the First International Randomized trial in advanced or Metastatic Adrenocortical Carcinoma Treatment (FIRM-ACT
Table 3. Mitotane dose regimen, glucocorticoid, and mineralocorticoid supplementation, blood level monitoring, and dose adjustment according to toxicity and blood level monitoring
Mitotane dose regimena • Start with 1.5 g/d and increase dose within 4–6 days to 6 g/days
• After 3 weeks, adjust dosage according tolerability and blood level (see below)
• Maximum dose 12 g/days, but most patients do not tolerate >8 g/days
• Target mitotane blood level 14–20 mg/l. Using this regimen, ∼50% of patients achieve the target level within 3 months
Glucocorticoid and mineralocorticoid supplementation
• A total daily dose of 50 mg hydrocortisone (divided as 20–20–10 mg) or 75 mg cortisone acetate and more may be needed. Glucocorticoid replacement is monitored best with careful clinical assessment
• Fludrocortisone may be added depending on the blood pressure, serum potassium levels, and plasma renin activity
Recommended blood monitoring during mitotane therapy
• Mitotane serum levels every 2–3 weeks in the first 3 months. After reaching a plateau, the interval can be extended (i.e. every 6 weeks)
• Glutamate-Oxaloacetate Transaminase (GOT), Glutamate-Pyruvate Transaminase (GPT), bilirubin, Gamma-Glutamyl-Transferase (GGT). Initially every 4 weeks, after 6 months every 8 weeks. GGT is invariably elevated without clinical consequences. If other liver enzymes are rapidly increasing (>3-fold of baseline), there is a risk of liver failure: stop mitotane
• TSH, fT3, fT4 every 3–4 months. Thyroid hormone replacement is recommended in patients with clinical symptoms of hypothyroidism
• Testosterone, free testosterone, and sexual hormone binding globulin (SHBG) should be tested in male patients with symptoms of hypogonadism
• Renin every 3 months. If renin increases in the presence of symptoms suggestive of mineralocorticoid deficiency, fludrocortisones should be added
• Cholesterol (High-Density Lipoprotein (HDL), Low-Density Lipoprotein (LDL)), triglycerides every 3– 4 months (in an adjuvant setting). If LDL/HDL cholesterol consistently increases, consider treatment with statins not metabolized by CYP3A4 (e.g. pravastatin, rosuvastatin)
• Blood count every 3–4 months
Plasma mitotane level CNS (grade 2)/GI side effects (grade 3/4) Grade 3/4 CNS side effects
Absent Present Present
<14 mg/l Increase daily dose by 1 gb Reduce daily dose by 1 g Stop mitotanec
14–20 mg/l Maintain dose Reduce daily dose by 1.5 g Stop mitotanec
>20 mg/l Reduce daily dose to 50%–75% of the most recent dose
Stop mitotanec Stop mitotanec
Recommended dose adjustment according to the central nervous system (CNS)/gastrointestinal (GI) side effects and plasma mitotane level. HDL, high-density lipoprotein; LDL, low-high-density lipoprotein; GOT, glutamate-oxaloacetate transaminase; GPT, glutamate-pyruvate transaminase; GGT, gamma-glutamyl-transferase.
aAn alternative low-dose regimen is also available with potentially similar efficacy. bup to the maximum tolerated dose.
c
Until symptom resolution (grade 0 or 1)
trial), the two most active treatment regimens namely
Etoposide, Doxorubicin, Cisplatin, and Mitotane (EDP-M) and Streptozotocin and Mitotane (Sz-M) were compared with 304 chemotherapy-naïve patients. Patients with disease progression received the alternate regimen. The results of this trial, recently published [26], indicate to us that EDP-M is the superior regimen.
Although no statistically substantial increase in overall survival was documented in patients receiving EDP-M as the first-line therapy, significantly better response rates and progression-free survival rates were achieved with EDP-M in comparison with Sz-M. The rate of serious adverse events was comparable. Of note, the results of the second-line regimens replicated the rates observed with thefirst-line therapy. Since EDP-M was superior to Sz-M in terms of progression-free survival either asfirst-line or second-line therapy, the crossover design may have attenuated its advantage on overall survival. On these bases, we recommend EDP-M as thefirst-line therapy for ACC requiring cytotoxic therapy, and as the reference for new therapies (I, A; Figure1A).
In patients unfit for the EDP-M regimen P-M may constitute a reasonable alternative [27]. However, the FIRM-ACT trial with a median overall survival between 12 and 14.8 months clearly indicated that new systemic therapy options are urgently needed, but thus far positive results are lacking despite an increasing number of prospective studies, also incorporating modern targeted agents. Results of studies with oral CYP3A4-mediated drugs may have been hampered by subtherapeutic systemic exposure due to CYP3A4 induction by mitotane [28,29]. A multicenter prospective randomized, placebo-controlled clinical trial aimed to test the efficacy of OSI 906–301, an IGR inhibitor, as second-/third-line approach in ACC patients has recently completed patient accrual. However, the results of this study will not be available before 2013.
In case of painful metastasis, palliative radiotherapy is an option, particularly in bone lesions (IV, B). Arterial chemoembolization and radiofrequency ablation may be beneficial in selected patients (V, C).
The therapeutic strategy of metastatic pheochromocytoma/ paraganglioma aims to control excessive catecholamine secretion and tumor burden, but no curative treatment is achievable. Treatment choices include a wait and see policy, locoregional therapies, systemic chemotherapy, and
radiopharmaceutical agents (Figure1B) [2], and they should be discussed case by case in a multidisciplinary specialized setting. In the absence of any randomized studies and demonstrated impact on survival, the patients’ quality of life should always be considered. Indeed, due to the indolent course of subgroups of patients, a wait and see policy coupled with a watchful follow-up can be considered as an option in asymptomatic patients with a low tumor burden. In these patients, an antineoplastic treatment should be recommended in case of rapid progression and/or symptom onset. In the absence of tumor progression, surgery of the primary tumor or metastases can reduce hormone secretion and may prevent complications related to a critical anatomical location and improve the efficacy of subsequent therapies [2]. Metastatic disease palliation may also benefit from local therapy with embolization and or
radiofrequency ablation. Radionuclide therapy is an effective treatment and131I-MIBG, in activities ranging from 5.5 to 38 GBq (150–1000 mCi), is the most frequently used.
Approximately 50% of patients are eligible for131I-MIBG therapy based on the uptake on diagnostic scans. Several studies have been published on the efficacy of131I-MIBG treatment [2]; most are retrospective, and only one is a prospective phase II study [30]. Objective responses were observed in 22–47% of cases. Long-term survival of responders of 4.7 years or 72 months was reported but progression at study entry was not a prerequisite of most studies. Objective responses were mainly observed in patients with soft tissue metastases. Grade 3–4 toxicity was reported in 16%–83% of patients.
131
I-MIBG therapy should be considered as afirst-line approach in patients with a good uptake of123I-MIBG and unresectable, progressive pheochromocytoma/paraganglioma or symptomatic patients (not amenable to locoregional control), or patients with a high tumor burden with a low number of bone metastases. More recently peptide-radiolabeled radiotherapy has also been developed.
Cyclophosphamide- and dacarbazine-based regimens combined with vincristine (CVD) or doxorubicin (CVDD or CDD) are the best studied chemotherapy regimens [2]. In the largest published study to date (n = 52 patients), 40% of patients treated with CVD, CDD, or CVDD experienced clinical benefits, including a reduction in tumor size in 25% of cases [31]. Systemic chemotherapy is debated as afirst-line therapy in patients with a low uptake of123I-MIBG and unresectable, rapidly progressive pheochromocytoma/ paraganglioma, or patients with high tumor burden or with a high number of bone metastases. As in well-differentiated pancreatic neuroendocrine tumors, there is a strong rationale and somefirst evidence on the potential efficacy of anti-angiogenic drugs in malignant pheochromocytomas. Thus, the European ENSAT network is currently launching a
randomized, placebo-controlled trial testing sunitinib in patients with malignant pheochromocytoma and paraganglioma (FIRST-MAPPP trial).
follow-up
International recommendations on follow-up are lacking both for ACC and pheochromocytoma/paraganglioma, and this is a significant problem. The recommendations in these guidelines were formulated based on data available on the natural history of these diseases, personal experience, and consensus among panelists.
For patients with ACC after complete resection, we recommend regular follow-up every 3 months including abdominal CT (or MRI), thoracic CT, and monitoring of initially elevated steroids (V, B). After 2 years, intervals may be gradually increased. In case of long-term persistence of the disease-free status, follow-up should be continued for at least 10 years.
For locally advanced or metastatic disease, overall survival and time to progression (TTP) are the most important endpoints with the response rate (RR) as a secondary end point. The TTP and RR guide the clinical decision-making in
an individual patient and should be evaluated at regular intervals (every 12 weeks or less depending on the therapy) using CT scans. The role of PET scans is not yet clear in ACC.
Patients who underwent successful surgery for non-metastatic pheochromocytoma/paraganglioma are at risk of
malignant recurrence and require long-term clinical (adrenergic symptoms and blood pressure levels) and biochemical ( plasma or urinary metanephrine,
normetanephrine, chromogranin A, and methoxythyramine) follow-up [2]. The follow-up is especially important for patients with extra-adrenal primary disease, tumor size >5 cm, or SDHB mutations. Biochemical testing is repeated∼14 days following surgery to check for remaining disease and thereafter every 3–4 months for 2–3 years. This should subsequently be repeated every 6 months. Patients with new events (high blood pressure, adrenergic symptoms, or pain) or pathological endocrine tests and/or elevated circulating chromogranin A should undergo imaging that includes thorax and abdomen CT and best functioning imaging (PET FDG in most cases). In case of proven malignant disease, SDHB mutation, extra-adrenal primary disease, and in rare cases of
pheochromocytoma/paraganglioma without relevant
preoperative hormone secretion, imaging could be repeated at least every 6 months during thefirst year and yearly afterward, irrespective of the negative results of biochemical tests. In these patients, lifelong follow-up is recommended [2].
con
flict of interest
Dr. Berruti has reported: advisory board honorarium from Astellas. Prof. Fassnacht has reported Principal Investigator of a study funded by HRA Pharma, Research grants: Pfizer, Astellas Pharma.
The other authors have reported no potential conflicts of interest.
references
1. Fassnacht M, Libé R, Kroiss M et al. Adrenocortical carcinoma: a clinician’s update. Nat Rev Endocrinol 2011; 7: 323–335.
2. Pacak K, Eisenhofer G, Ahlman H et al. International symposium on pheochromocytoma. Pheochromocytoma: recommendations for clinical practice from the first international symposium. October 2005. Nat Clin Pract Endocrinol Me tab 2007; 3: 92–102.
3. Bertherat J, Bertagna X. Pathogenesis of adrenocortical cancer. Best Pract Res Clin Endocrinol Metab 2009; 23: 261–271.
4. Esenhofer G, Lenders JW, Siegert G et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer 2012; 48: 1739–1749.
5. Arlt W, Biehl M, Taylor AE et al. Urine steroid metabolomics as a biomarker tool for detecting malignancy in adrenal tumors. J Clin Endocrinol Metab 2011; 96: 3775–3784.
6. Timmers HJ, Kozupa A, Chen CC et al. Superiority offluorodeoxyglucose positron emission tomography to other functional imaging techniques in the evaluation of metastatic SDHB-associated pheochromocytoma and paraganglioma. J Clin Oncol 2007; 25: 2262–2269.
7. Sbiera S, Schmull S, Assie G et al. High diagnostic and prognostic value of steroidogenic factor-1 expression in adrenal tumors. J Clin Endocrinol Metab 2010; 95: E161–E171.
8. Weiss LM, Medeiros LJ, Vickery AL, Jr. Pathologic features of prognostic significance in adrenocortical carcinoma. Am J Surg Pathol 1989; 13: 202–206. 9. Fassnacht M, Johanssen S, Quinkler M et al. German adrenocortical carcinoma
registry group; European Network for the Study of Adrenal Tumors. Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: proposal for a Revised TNM Classification. Cancer 2009; 115: 243–250.
Table 4. Summary of Recommendations
• ACC is defined by a Weiss score of 3 or more. Malignant
pheochromocytomas/paragangliomas are defined by the presence of metastasis.
• Patients suspected to harbor primary adrenal tumors should undergo a standardized diagnostic work-up consisting of endocrine assessment for excess hormone production and modern imaging (CT/MRI of abdomen, chest CT, and in selected cases supplemented by isotope functional imaging mainly FDG-PET). The diagnostic work-up differs between ACC and pheochromocytoma.
• Guided biopsies of potentially resectable primary adrenal tumors are not informative in most cases, but are potentially harmful and should be avoided.
• The ENSAT TNM staging system should be used for ACC staging.
• Histological diagnosis should be done by an experienced pathologist and should rely on morphological, mitotic, and immunohistochemical parameters.
• Complete surgical extirpation of localized and locally advanced ACC or pheochromocytoma (R0 resection) is the mainstay of potentially curative approaches. Additionally, a locoregional lymphadenectomy is suggested for ACC.
• In pheochromocytoma cytoreductive surgery might be considered. In advanced ACC, this approach is only reasonable for patients with severe hormone excess.
• Meticulous perioperative management of hormonal, glucose, electrolytes, cardiac andfluid/blood pressure abnormalities is a critical component of patient care.
• Despite the limited literature evidence, adjuvant systemic mitotane is recommended for patients with ACC and incomplete resection (R1, Rx stage III) or in the presence of high-risk features (Ki67>10%). R1 and Rx ACC resections may be followed by additional adjuvant radiotherapy to the tumor bed.
• Fit patients with inoperable ACC, high tumor volume and rapid disease progression should be treated with combination cytotoxic chemotherapy plus mitotane (EDP-M). Lessfit patients and/or patients with low tumor burden and slow progression can (first) be managed with mitotane monotherapy combined or not with locoregional options.
• Disease and symptom control is the main treatment goal for patients with inoperable pheochromocytoma and can be attempted by radiopharmaceuticals (131I-MIBG), locoregional ablative procedures, and/or combination chemotherapy (CVD) in selected cases.
• Wait and see policy is recommended in low tumor burden and asymptomatic malignant pheochromocytoma and paraganglioma.
• Patients with resected ACC or pheochromocytoma should be followed at regular intervals with clinical, imaging and biochemical screens for at least 10 years. Lifelong surveillance with an increased interval of time is favored in malignant pheochromocytoma/paraganglioma.
• The follow-up of patients with inoperable disease should be performed every 2–4 months for ACC and every 3–6 months for
pheochromocytoma/paraganglioma during thefirst year of follow-up and then adjusted.
10. Baudin E, Leboulleux S, Al Ghuzlan A et al. Therapeutic management of advanced adrenocortical carcinoma: what do we know in 2011? Horm Cancer 2011; 6: 363–371.
11. de Reyniès A, Assié G, Rickman DS et al. Gene expression profiling reveals a new classification of adrenocortical tumors and identifies molecular predictors of malignancy and survival. J Clin Oncol 2009; 27: 1108–1115.
12. Ayala-Ramirez M, Feng L, Johnson MM et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: primary tumor size and primary tumor location as prognostic indicators. J Clin Endocrinol Metab 2011; 96: 717–725.
13. Amar L, Baudin E, Burnichon N et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab 2007; 92: 3822–3828. 14. Lombardi CP, Raffaelli M, Boniardi M et al. Adrenocortical carcinoma: effect of
hospital volume on patient outcome. Langenbecks Arch Surg 2012; 397: 201–207. 15. Porpiglia F, Fiori C, Daffara F et al. Retrospective evaluation of the outcome of
open versus laparoscopic adrenalectomy for stage I and II adrenocortical cancer. Eur Urol 2010; 57: 873–878.
16. Icard P, Goudet P, Charpenay C et al. Adrenocortical carcinomas: surgical trends and results of a 253-patient series from the French Association of Endocrine Surgeons study group. World J Surg 2001; 25: 891–897.
17. Reibetanz J, Jurowich C, Erdogan I et al. Impact of lymphadenectomy on the oncologic outcome of patients with adrenocortical carcinoma. Ann Surg 2012; 255: 363–369.
18. Schteingart DE, Doherty GM, Gauger PG et al. Management of patients with adrenal cancer: recommendations of an international consensus conference. Endocr Relat Cancer 2005; 12: 667–680.
19. Pacak K. Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab 2007; 92: 4069–4079.
20. Amar L, Servais A, Gimenez-Roqueplo AP et al. Year of diagnosis, features at presentation, and risk of recurrence in patients with pheochromocytoma or secreting paraganglioma. J Clin Endocrinol Metab 2005; 90: 2110–2116.
21. Polat B, Fassnacht M, Pfreundner L et al. Radiotherapy in adrenocortical carcinoma. Cancer 2009; 115: 2816–2823.
22. Terzolo M, Angeli A, Fassnacht M et al. Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med 2007; 356: 2372–2380. 23. Berruti A, Fassnacht M, Baudin E et al. Adjuvant therapy in patients with
adrenocortical carcinoma: a position of an international panel. J Clin Oncol 2010; 28: e401–e402.
24. Haak HR, Hermans J, van de Velde CJ et al. Optimal treatment of adrenocortical carcinoma with mitotane: results in a consecutive series of 96 patients. Br J Cancer 1994; 69: 947–951.
25. Hermsen IG, Fassnacht M, Terzolo M et al. Plasma concentrations of o,p’DDD, o, p’DDA, and o,p’DDE as predictors of tumor response to mitotane in
adrenocortical carcinoma: results of a retrospective ENS@T multicenter study. J Clin Endocrinol Metab 2011; 96: 1844–1851.
26. Fassnacht M, Terzolo M, Allolio B et al. Combination chemotherapy in advanced adrenocortical carcinoma. N Engl J Med 2012; 366: 2189–2197.
27. Bukowski RM, Wolfe M, Levine HS et al. Phase II trial of mitotane and cisplatin in patients with adrenal carcinoma: a Southwest Oncology Group study. J Clin Oncol 1993; 11: 161–165.
28. van Erp NP, Guchelaar HJ, Ploeger BA et al. Mitotane has a strong and a durable inducing effect on CYP3A4 activity. Eur J Endocrinol 2011; 164: 621–626.
29. Ayala-Ramirez M, Feng L, Habra MA et al. Clinical benefits of systemic chemotherapy for patients with metastatic pheochromocytomas or sympathetic 675 extra-adrenal paragangliomas: insights from the largest single-institutional experience. Cancer 2012; 118: 2804–2812.
30. Kroiss M, Quinkler M, Lutz WK et al. Drug interactions with mitotane by induction of CYP3A4 metabolism in the clinical management of adrenocortical carcinoma. 665 Clin Endocrinol (Oxf ) 2011; 75(5): 585–591.
31. Gonias S, Goldsby R, Matthay KK et al. Phase II study of high-dose [131I] metaiodobenzylguanidine therapy for patients with metastatic pheochromocytoma and paraganglioma. J Clin Oncol 2009; 27: 4162–4168.