HAL Id: hal-03160951
https://hal.sorbonne-universite.fr/hal-03160951
Submitted on 5 Mar 2021
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
phase IIa trial (Lupsenic)
Mohamed Hamidou, Antoine Néel, Joel Poupon, Zahir Amoura, Mikael Ebbo,
Jean Sibilia, Jean-Francois Viallard, Benjamin Gaborit, Christelle Volteau,
Jean Benoit Hardouin, et al.
To cite this version:
Mohamed Hamidou, Antoine Néel, Joel Poupon, Zahir Amoura, Mikael Ebbo, et al.. Safety and
efficacy of low-dose intravenous arsenic trioxide in systemic lupus erythematosus: an open-label phase
IIa trial (Lupsenic). Arthritis Research & Therapy, BMC, 2021, 23 (1), pp.70.
�10.1186/s13075-021-02454-6�. �hal-03160951�
R E S E A R C H A R T I C L E
Open Access
Safety and efficacy of low-dose intravenous
arsenic trioxide in systemic lupus
erythematosus: an open-label phase IIa trial
(Lupsenic)
Mohamed Hamidou
1*†, Antoine Néel
1†, Joel Poupon
2, Zahir Amoura
3, Mikael Ebbo
4, Jean Sibilia
5,
Jean-Francois Viallard
6, Benjamin Gaborit
1, Christelle Volteau
7, Jean Benoit Hardouin
8, Eric Hachulla
9and
François Rieger
10Abstract
Background: Lupus animal model has shown that arsenic trioxide (ATO), a treatment of acute promyelocytic leukaemia, could be effective in SLE. This is the first clinical study to determine the safety and efficacy of a short course of intravenous ATO in patients with active SLE.
Methods: This phase IIa, open-label, dose-escalating study enrolled 11 adult SLE patients with a non-organ threatening disease, clinically active despite conventional therapy. Patients received 10 IV infusions of ATO within 24 days. The first group received 0.10 mg/kg per injection, with dose-escalating to 0.15 mg/kg in a second group, and to 0.20 mg/kg in a third group. The primary endpoint was the occurrence of adverse events (AEs) and secondary endpoints were the number of SLE Responder Index 4 (SRI-4) responders at week 24 and reduction of corticosteroid dosage. In an exploratory analysis, we collected long-term data for safety and attainment of lupus low disease activity state (LLDAS).
Results: Four serious AEs occurred (grade 3 neutropenia, osteitis, neuropathy), 2 of which were attributable to ATO (neutropenia in the 2 patients treated with mycophenolate). Two patients suffered a severe flare during the last 4 weeks of the trial. At W24, five patients among 10 were SRI-4 responders. Overall, mean corticosteroid dosage decreased from 11.25 mg/day at baseline to 6 mg/day at W24 (P < 0.01). In the long term, 6 patients attained LLDAS at W52, which continued at last follow-up (median LLDAS duration 3 years, range 2–4).
Conclusions: A short course of ATO has an acceptable safety profile in SLE patients and encouraging efficacy. Trial registration: ClinicalTrials.gov,NCT01738360registered 30 November 2012
Keywords: Systemic lupus erythematosus, Autoimmune diseases, Treatment, Arsenic trioxide, Phase II clinical trial
© The Author(s). 2021 Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visithttp://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
* Correspondence:mohamed.hamidou@chu-nantes.fr
†Mohamed Hamidou and Antoine Néel equally contributed as co-first
authors.
1Department of Internal Medicine, CHU Nantes, Nantes Université, Nantes,
France
Background
Systemic lupus erythematosus (SLE) is a heterogeneous autoimmune disease characterised by overproduction of type I interferons, hyperactivity of B cells with production of antibodies, presence of autoreactive T cells and activa-tion of NETosis [1]. In SLE patients with persistent disease activity despite antimalarials and low dose steroids, quality of life is adversely affected by the unpredictable risk of flares and by drug toxicity and accumulation of organ damage. Belimumab is the only treatment approved since 2011 in refractory mild to moderate SLE, especially with high activity and positivity of biomarkers [2]. Currently, many controlled phase II/III trials evaluating targeted therapies have failed to meet their primary end point [3]. Drugs primarily used in onco-haematology may exert targeted or pleiotropic immunological effects and thus be effective in SLE. Unfortunately, phase III clinical trials evaluating rituximab failed to meet their primary end point [4, 5]. More recently, bortezomib, a proteasome inhibitor approved in multiple myeloma, demonstrated unacceptable toxicity in 12 SLE patients [6]. Trials evalu-ating new drugs such as Janus kinase or Bruton’s tyrosine kinase inhibitors are ongoing.
Arsenic trioxide (ATO), As2O3has been approved for
the treatment of acute promyelocytic leukaemia (APL) [7, 8], and more than 100 trials involving ATO are ongoing for treatment of solid and haematological neo-plasias (myeloma, B cell neoplasia, etc.), in adults, as well as in elderly patients and children [9–11]. In recent years, ATO has been studied in several animal models of SLE [12, 13], systemic scleroderma [14] and chronic graft-versus-host disease (cGVHD) with scleroderma-like features [15]. Bobé et al. [12] demonstrated striking results with intraperitoneal injection of ATO treatment in the murine lupus model MRL-lpr/lpr, characterised by an autoimmune lymphoproliferative syndrome, with tissue infiltration by double-negative T lymphocytes, and lupus-like manifestations. As2O3 induced a rapid and marked decrease in the size of spleen and lymph nodes and suppressed skin lesions compared to PBS treated control MRL/lpr mice. It also inhibited auto-antibody production and immune complex deposition in the kidney and prevented nephritis. Furthermore, arsenic drastically improved the survival of treated mice. In this model, ATO did not act as a cytotoxic immunosuppres-sant. Rather, it exerted a selective pro-apoptotic effect of deletion of autoreactive lymphocytes, inhibited the production of autoantibodies, and reduced the levels of cytokines. In peripheral blood mononuclear cells of lupus patients, ATO reduces the expression level of IFN-gamma [16].
Thus, given the promising results in preclinical studies, we conducted this pilot open-label, dose-escalating multi-centre phase IIa trial, to evaluate the safety and efficacy of
a short course of ATO in the treatment of active SLE with inadequate responses to standard therapy.
Methods
Study design and patients
LUPSENIC was a phase IIa, open-label controlled, multicen-tre trial, evaluating dose-escalation intravenous ATO in SLE (EudraCT: 2012-002259-40; NCT: NCT01738360). The study was conducted between July 2013 and October 2015 and included patients from 6 university hospitals in France (Nantes, Lille, Paris, Bordeaux, Marseille, Strasbourg). This pilot trial was designed to examine the safety profile and tolerability of ATO as a primary objective, with efficacy as a secondary objective, in patients with active SLE.
All patients fulfilled at least four of the 11 American College of Rheumatology (ACR) criteria for SLE [17,18], were aged 18 years or more and had a clinically active disease, defined by a Safety of Estrogens in Lupus Erythematosus: National Assessment–SLE Disease Activity Index (SELENA-SLEDAI) score > 6 [19], despite standard-of-care treatment. All patients were seropositive for anti-nuclear antibody (ANA) (≥ 1/80 in immunofluor-escence assay). Patients with severe active renal or CNS disease, a history of cardiac disease, cytopenia with haemoglobin < 11 g/dl, neutrophil count < 1200/m3, and/ or platelet count < 100,000/ml were excluded. Patients had to be receiving prednisone dose ≥ 10 mg/day and be refractory, for at least 1 year, or intolerant, to hydroxy-chloroquine (HCQ). Concomitant therapy with HCQ and/ or immunosuppressive drugs (methotrexate [MTX], azathioprine [AZA], mycophenolate mofetil [MMF]) were allowed at stable doses for a minimum of 6 months before the start of study treatment Corticosteroid dosage tapering was left to investigators’ discretion, based on their clinical judgement.
The therapeutic scheme was designed on the basis of pharmacokinetics data obtained from APL treatments in previous studies: [20] patients were administered a loading dose of ATO on each of the first 4 days (as in-patients), then twice a week (as outpatients) during weeks 2 to 4, i.e. 10 IV infusions (duration: 4 h) within 24 days. To avoid the risk of QT prolongation, patients had to have normal kaliema and magnesemia at the start of, and during, the treatment period. Dose escalation was conducted using the continual reassessment method [21, 22]. The ATO doses were studied sequentially, patients receiving 0.1 mg/kg per injection, 0.15 mg/kg per injection or 0.20 mg/kg per injection (see online supplementary S1and S2).
All patients provided written informed consent in accordance with the declaration of Helsinki. The study was approved by regional Human Subjects Research Protection Committee (Comité de Protection des Personnes Ouest-I, Tours hospital). All the safety data were submitted
to an independent Data and Safety Monitoring Board (DSMB).
Assessments
The primary endpoint was the occurrence of adverse events (AEs) and the secondary endpoints evaluated the efficacy of ATO. During the 24 weeks of the trial, patients made regular scheduled visits, at baseline and at weeks 8, 12, 16, 20, and 25. The last patient visit oc-curred in October 2015. Long-term follow-up data were updated in February 2018, in terms of safety (severe AE [SAE], death) and efficacy (lupus low disease activity state [LLDAS], severe flare, corticosteroid dosage).
Primary and secondary outcome measures
The primary outcome measures for safety were the re-cording of AEs evaluated by NCI Common Terminology Criteria, including incidence, nature and severity of AEs, clinical laboratory abnormalities, vital signs, ECGs and physical examination findings. Serum creatinine, potas-sium and magnepotas-sium, complete blood cell count and transaminases were checked before each infusion of ATO and at each visit. The main secondary outcome was the efficacy of ATO on disease activity. Disease activity was evaluated using the SELENA-SLEDAI [19], the British Isles Lupus Assessment Group (BILAG) index [23], the physician’s global assessment (PGA) and
clinical laboratory assessments, including proteinuria and serum biomarkers (anti-dsDNA antibodies, C3 and C4 complement, total IgG, IgA IgM dosage). Testing for anti-dsDNA antibodies was performed by Elisa or Farr test, according to local standard practice, using a single method for each patient.
Flares were defined according to the SELENA-SLEDAI Flare Index (SFI): [24, 25] flares were mild or moderate if changes in the SELENA-SLEDAI score were > 3 points or severe if the SELENA-SLEDAI score increased by 12 points. SELENA-SLEDAI, PGA and BILAG index scores and corticosteroid dosage were recorded every 4 weeks, as were AEs, vital signs, concomitant medications and laboratory results.
We assessed the SRI-4 response at week 24. SRI-4 re-sponders were defined as having a reduction of at least 4 points in the SELENA-SLEDAI score, no new BILAG A or no more than one new BILAG B and no worsening in their PGA score [26]. Health-related quality of life was assessed with the 36-item Short-Form Health Survey (SF-36). Corticosteroid sparing was defined as a reduc-tion in oral corticosteroid (OCS) dosage to 7.5 mg/day or less.
Arsenic level was determined in serum and erythro-cytes by inductively coupled plasma-mass spectrometry before and after each IV infusion.
Long term follow-up
In a long term follow-up analysis, we evaluated the number of patients who attained LLDAS within 52 weeks after ATO administration. LLDAS was defined as follows: SELENA-SLEDAI score≤ 4 without major organ activity, no new disease activity, no new features of lupus disease activity compared with the previous assessment, PGA ≤ 1 on a 3-point scale, current prednisolone-equivalent dosage < 7.5 mg/d and well-tolerated standard maintenance dosages of immunosuppressive drugs and approved biologics [27]. We also evaluated time to first flare (according to SFI and BILAG) beyond W24 for SRI responders and patients who attained LLDAS at W52. Statistical analysis
Descriptive statistics are presented as median and inter-quartile range (IQR) for quantitative variables, or counts and percentages for categorical variables, unless other-wise indicated. No formal statistical calculation was performed to determine the necessary sample size. The continual reassessment method was performed with R v.3.1.1. The statistical significance of changes in SLEDAI, daily OCS dose and PGA compared to baseline values was tested using mixed model for repeated measure-ments. Analyses were performed with SAS v.9.
Results
Study population
Eleven patients were included in the study. All were Caucasians, eight were women and the mean age was 44 years. At entry in the study, all patients had active SLE in the mucocutaneous and/or musculoskeletal domain, were treated with OCS at doses ≥ 10 mg/day and anti-malarials (except 1 patient with a history of APS-related maculopathy). Six among 11 had a concomitant im-munosuppressive treatment (3 MTX, 2 MMF and 1 AZA). All patients had ANA ≥ 1/80, five had low C3 complement levels, three had low C4 levels and six had anti-dsDNA antibodies.
Four patients received 0.10 mg/kg per injection of ATO, four patients had 0.15 mg/kg per injection and three patients had 0.20 mg/kg per injection. Among the 11 included patients, one patient was excluded at day 17, from the study and from the dose escalation calculation with DSMB approval, because of protocol violation (administration of ATO despite grade 3 neutropenia). Safety profile
All the patients were evaluated for safety analysis. No dose-limiting toxicity (DLT) occurred and the study was discontinued when we reached 11 ATO-treated patients. Among the 11 included patients, two patients discontinued ATO after seven injections because of treatment-related SAEs. A description of AEs by System Organ Class is given
in Table1. Eighteen AEs were considered by the treating physicians to be related to the study drug and were gener-ally mild to moderate. One patient had moderate hypomag-nesemia and hypokaliema, five patients had diarrhoea and two patients had asymptomatic moderate QT prolongation (< 470 msec). Four SAEs occurred in four patients during the study, and two were formally considered by the treating physicians to be ATO-related. The latter occurred in two patients treated with ATO 0.15 and 0.20 mg/kg, respect-ively, and also receiving MMF, who experienced asymp-tomatic transient grade 3 neutropenia (neutrophil nadir at 0.69 and 0.51 G/L) leading to permanent discontinuation of ATO. Both patients showed complete spontaneous recov-ery in less than 5 days, without infection. There was no clear correlation between the arsenic levels reached and the observed toxicity. Another patient (ATO 0.2 mg/kg), had a grade 2 reversible sensory peripheral neuropathy, con-comitant with acute hepatitis A virus infection. One patient (ATO 0.15 mg/kg) treated with a non-steroidal anti-inflammatory drug and 10 mg/day of prednisone had chronic cutaneous ulceration of a toe (Jaccoud arthropathy), which was complicated by an osteitis, requiring limited amputation of the distal phalanx.
Efficacy Clinical efficacy
As mentioned previously, one patient was excluded due to protocol violation. Among 10 patients assessed for efficacy, nine patients received 10 ATO IV injections as planned in the protocol, and one patient discontinued ATO after seven injections, because of neutropenia re-lated to ATO.
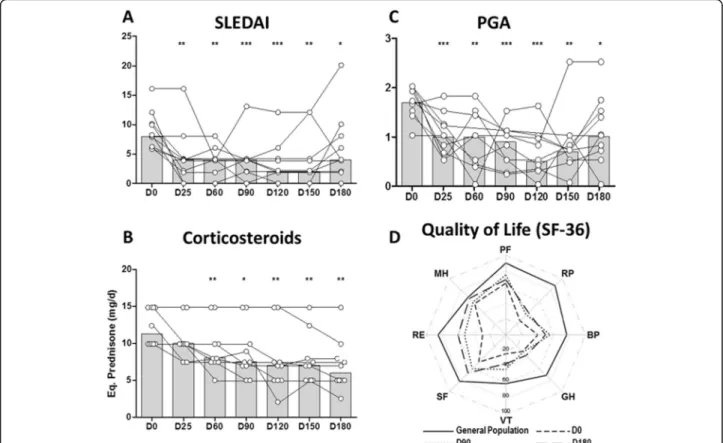
Five out of ten patients (50%) achieved a SRI-4 re-sponse at W24, three with 0.1 mg/kg ATO dosage and two with 0.20 mg/kg ATO dosage (Table 2). Median SLEDAI score at inclusion was 8 and decreased to 3.4 points at week 16 (Fig.1a, p < 0.001). Two patients had a severe flare, one at W24 and the other W20, with BILAG A respectively in the renal and mucocutaneous domains, respectively. They received immunosuppres-sant after W24. Two other patients had BILAG B in the mucocutaneous domains. There were no significant dif-ferences in SRI-4 responder rates among the three ATO regimens.
Tapering of OCS was done at the treating physician’s discretion. The median dosage of prednisone decreased from 12.25 mg/day at baseline to 6 mg/day at W24 (Fig. 1b, p = 0.0046). At W24, seven out of 10 patients reached the 7.5 mg/day target-dose.
Biomarkers and ATO pharmacokinetics
Among the six patients with detectable anti-dsDNA antibodies at baseline, a trend towards an initial decrease was observed from W4 to W8 (Fig. 2a). Levels of C3, C4, and IgG were not modified (Fig. 2b and c); levels of IgA and IgM were not modified (data not shown). Ro/ SSA and anticardiolipin antibodies likewise remained unchanged (data not shown). Plasma arsenic concentra-tions were measured just before (residual) and at the end of the infusion for the three groups of ATO dosage (Fig. 3). During the induction phase (D1-D4), we ob-served a regular increase in arsenic concentrations in the three groups proportionally to the amount of ATO injected. At D8, arsenic concentrations decreased and stayed relatively stable between D8 and D25, with, large inter-individual variations after D8, especially in the 0.15 mg/kg group. Pharmacokinetic data did not show any correlation between plasma ATO levels and safety and efficacy (data not shown).
Long-term follow-up
Long-term follow-up data are reported in Table 3. One year after ATO administration, six out of 10 patients attained LLDAS with no further therapeutic intervention, and this state persisted at the last follow-up (median dur-ation 45 months, range 34–51). At last follow-up, median OCS dosage of these patients was 2.8 mg/day (range 0–5). In terms of safety, no SAE related to ATO treatment was Table 1 Arsenic trioxide safety at week 24
Adverse event type n Total number of ATO-related AEs 22 Treatment-emergent SAEs 4
Neutropenia* 2
Axonal neuropathy† 1
Osteitis‡ 1
AEs leading to ATO discontinuation
Neutropenia grade 3 2 Treatment-related AEs (mild to moderate) 18
Anaemia (grade 2) 2 Diarrhoea (grade 1) 5
Nausea 2
Hypomagnesaemia 1 Mild QT prolongation (grade 1) 2 Injection-related reaction 1 Injection site reaction 1
Hypotension 1
Flushing 1
Increased transaminases (grade 1) 1
Fatigue 1
* Neutropenia in 2 patients treated respectively with 0.15 and 0.20 mg/kg of ATO associated with MMF
†Neuropathy associated with acute hepatitis A virus infection ‡Osteitis of a toe in a patient with Jaccoud arthropathy
AEs adverse events, SAE severe adverse event, ATO arsenic trioxide, MMF mycophenolate mofetil
Table 2 Patients baseline characteristics and ATO efficacy at week 24 Baselin e AT O dos age m g/kg/d We ek 24 Patie nt no., gender, age (year s) SLE dur ation (year s) Prev ious IS Con comitan t trea tment Sympt oms SLE DAI OCS mg/d Severe flare SLE DAI OCS mg/d W 24 SRI-4 -R 1. F, 35 12 MTX HCQ, THA L, MTX A, C 10 15 0.1 0 W24 8 10 N o 2. F, 41 23 MTX HCQ A, C 12 15 0.1 0 No 2 2.5 Yes 3. F, 49 29 an ti-Fcg-R mAb HCQ A, C 10 15 0.1 0 No 4 7 Yes 4. M, 37 2 MTX HCQ, MTX A, C 6 10 0.1 0 No 2 8 Yes 5. F, 47 26 CYC , Ci, MMF, RT X, THAL HCQ A 8 15 0.1 5 No 10 15 N o 6. F, 30 4 BE LI, MT X, EPRA, LEFLU, HCQ A, C 6 10 0.1 5 No 6 7.5 N o 7. F, 50 14 MTX HCQ A 8 12.5 0.1 5 W20 20 2 N o 8. H, 53 3 MTX HCQ, MTX A, C 8 10 0.2 0 No 0 5 Yes 9. F, 45 15 MTX, AZ A, CYC MMF A, C 16 10 0.2 0 No 4 5 Yes 10. F, 23 6 AZ A HCQ, AZA A, C 6 10 0.2 0 No 4 5 N o ATO arsenic trioxide, AZA azathioprine, anti-Fcg-R mAb anti-Fc gamma-receptor monoclonal antibody, BELI belimumab, Ci ciclosporine, CYC cyclophosphamide, EPRA epratuzumab, HCQ hydroxychloroquin e, IS immunosuppressant, LEFLU leflunomide, MTX methotrexate, MMF mycophenolate mofetil, OCS oral corticosteroids, RTX rituximab, THAL thalidomide, A articular, C cutaneous, SLEDAI Systemic Lupus Erythematosus Disease Activity Index, SRI-4-R SLE response index-4 response
observed. A 34-year-old patient died from small-cell lung cancer 2 years after ATO administration. She was a to-bacco smoker and had received thalidomide and immuno-suppressive drugs for a long time, before and after ATO administration. ATO was deemed not to have been re-sponsible by the Pharmacovigilance Team.
Discussion
There is a significant unmet need for new agents for treating refractory SLE [3]. In this pilot study, we report the first-in-human assessment of the effect of ATO in an autoimmune disease, namely SLE. Our first finding is that a short course of ATO at a low dose demonstrated an acceptable safety and tolerability profile in active SLE patients, with skin and joint manifestations, steroid-dependant and unresponsive to HCQ and immunosup-pressive treatments.
In onco-haematology, ATO is administered at 0.15 mg/kg daily, for at least 5 weeks (i.e. 25 infusions), followed by maintenance therapy [27]. In our trial, pa-tients only received 10 infusions, without maintenance,
which resulted in a low total cumulated dose (1–2 mg/ kg). Interestingly, we noted that these low cumulative doses of ATO resulted in an encouraging rate of re-sponse in patients with steroid-dependent active SLE despite standard-of-care treatment. Five (50%) patients had a 24-week SRI-4 response. Seven among 10 patients could reduce corticosteroid dose below 7.5 mg/day. An improvement of quality of life was also observed. We noted a decrease in clinical SLEDAI score at W12, sug-gesting a rapid effect of ATO in our chosen regimen and its potential use as an induction treatment. In contrast and despite a trend towards a short-term decrease of anti-dsDNA antibodies, ATO treatment did not signifi-cantly modify complement or Ig levels.
Unexpectedly, a long-term follow-up update revealed that, in spite of the short-duration treatment and steroid tapering, 6 out of 10 patients achieved LLDAS within 52 weeks after ATO, which persisted at the last follow-up (Table3), up to 4 years after ATO administration. At last follow-up, 5 patients received ≤ 5 mg/day prednisone. LLDAS is a meaningful marker of response to treatment,
Fig. 1 Clinical efficacy outcomes of ATO. Efficacy results after ATO infusion. Treatment was given over 21 days and changes in a SLEDAI score, b corticosteroid dose and c PGA score were recorded every 4 weeks over 24 weeks thereafter. Histograms depict the median. *P < 0.05, **P < 0.01, ***P < 0.001 (mixed model for repeated measurements). d Median Health-Related Quality of Life (HRQoL) scores for the 8 dimensions of the SF36 for the patients at D0, D90 and D180 and the score of a gender- and age-matched sample of individuals drawn from the general French population. We noted a marked increase between D0 and D90/D180 for all the dimensions of the HRQoL and globally stable scores between D90 and D180, which remained inferior to those of the general population. ATO, arsenic trioxide; Eq., equivalent; PGA, Physician Global Assessment; SLEDAI, Systemic Lupus Erythematosus Disease Activity Index
correlated with a decrease in damage progression, and could be included in the assessment of future rando-mised controlled trials [28–31].
Four patients exhibited SAEs, only 2 of which were formally attributed to ATO, both corresponding to a
grade 3 neutropenia, reversible with ATO withdrawal. Neutropenia, an expected AE of ATO and MMF, occurred in the two patients concomitantly treated with MMF. Thus, we call for caution in the event of an association of ATO with MMF. Most of the other AEs
Fig. 2 Immunological effects of ATO. Changes in a anti-ds-DNA, b immunoglobulin G, c complement factor 3 and d complement factor 4 levels over 24 weeks after ATO infusion. Each dot represents an individual measurement. Changes in anti-dsDNA level are represented as variation from baseline in 6 patients with detectable anti-dsDNA at baseline. Dotted lines represent the lower limit of normal complement level
Fig. 3 ATO pharmacokinetics. Plasma arsenic concentrations (mean +/− SD) in the three groups of ATO dosage before (a) or at the end of the perfusion (b). White circle indicates 0.10 mg/kg, white square indicates 0.15 mg/kg, and black triangle indicates 0.20 mg/kg
Table 3 Long-term follow-up Patie nt
no., gender, age (year
s) AT O Dosa ge mg/k g/ d Week 24 Post-t rial lon g-term foll ow-up W24 trea tment S LEDAI score OCS mg/ day W2 4 S RI- 4-R W24 LLDA S W52 LLDA S Severe Flare At last foll ow-u p sustained LLDA S OCS mg/day LLDAS dur ation Trea tment 1. F, 35 0.10 HCQ, THA L, MTX 8 10 No No No W24, C No –– – 2. F, 41 0.10 HCQ 2 2.5 Yes Yes Yes No Yes 0 4y3 m HCQ 3. F, 49 0.10 HCQ 4 7 Yes Yes Yes No Yes 5 4y2 m HCQ 4. M, 37 0.10 HCQ, MTX 2 8 Yes No Yes No Yes 0 3y5 m HCQ, MT X 5. F, 47 0.15 HCQ 10 15 No No Yes No Yes 5 4y HCQ 6. F, 30 0.15 HCQ 6 7.5 No No Yes No Yes 7 2 y 10 m HCQ 7. F, 50 0.15 HCQ 20 2 No No No W20, R No –– – 8. H, 53 0.20 HCQ, MTX 0 5 Yes Yes Yes No Yes 0 3y6 m HCQ, MT X 9. F, 45 0.20 MMF 4 5 Yes Yes No W100 , R No –– – 10. F, 23 0.20 HCQ, AZA 4 5 No No No W80, R/H No –– – ATO arsenic trioxide, AZA azathioprine, HCQ hydroxychloroquine, MTX methotrexate, MMF mycophenolate mofetil, OCS oral corticosteroids, THAL thalidomide C, cutaneous, R renal, H heart, y years, m months, Lupus Low Disease Activity State, LLDAS SLEDAI, Systemic Lupus Erythematosus Disease Activity Index, SRI-4-R SLE response index-4 response
were mild and resolved after drug discontinuation. One 34-year-old patient developed small-cell lung cancer 2 years after discontinuation of ATO. She was a smoker, which remains the key risk factor for lung cancer in lupus patients [32]. In the onco-haematological setting, long-term follow-up of ATO-treated patients revealed no major chronic AEs, secondary carcinoma or arsenic retention [33].
During the daily ATO course (D0-D8), pharmacoki-netic analysis was consistent with data obtained in APL [19]. DLT was not reached and no dose-related trends in safety and efficacy were noted between the three dosages. Thus, a dosage of 0.15 mg/kg seems adapted for further studies in SLE and other systemic auto-immune diseases.
Data regarding the immunological effects of ATO are contradictory. In the treatment of APL, ATO has a specific pro-apoptotic effect on leukaemic cells and decreases the amount and inhibits the function of Treg cells [34]. However, in a murine islet allotransplantation model, ATO inhibited immune rejection and prolonged islet allograft survival by increasing the proportion of Foxp3+ Treg cells, inhibiting proliferation of T lympho-cytes and decreasing B lympholympho-cytes [35]. ATO, when combined with co-stimulation blockade, prolongs the survival of cardiac allografts in alloantigen-primed mice. In this model, splenic CD4(+) or CD8(+) memory T cells were significantly reduced, while the expression of Tregs was enhanced [36]. Arsenic has also been shown to re-duce dendritic cell cytokine production and the ability of dendritic cells to activate T cells in vitro [37]. Recently, Ye and coll. showed that As2O3 impaired activated pDCs of healthy donors to promote CD4 T cell prolifer-ation and Th1/Th22 polarizprolifer-ations. At clinically relevant concentrations, As2O3 inhibits preferentially IFN-a secretion, via IRF7 and plasmablast differentiation of B cells. They also observed on PBMC of untreated systemic sclerosis patients, that similarly to healthy pDCs, As2O3 induced preferential inhibition of IFN-a secretion, proa-poptotic effects and regulatory phenotype in SSc [38].
It is well established that dysregulation of oxidative stress is present in SLE [39]. In addition, ATO may be a potential immune modulator for treatment of rheuma-toid arthritis, that helps to balance of Treg and Th17 cells through modulating STAT3 [40]. Interestingly, an altered Th17/Treg balance is also present in SLE, and could be a target of ATO, through IL-17 [41].
The scheme tested herein as IV induction therapy (daily IV infusion during 4 days, then infusions twice a week until week 4) was well accepted by patients. In the future, oral ATO, currently used in APL [42, 43], could be a good option to test in autoimmune diseases.
The major limitations of our study are the small sam-ple size, the absence of a control group and the absence
of in-depth immunomonitoring, such as the type 1 inter-feron signature.
Conclusions
In this pilot clinical trial, a short course of intravenous ATO demonstrated an acceptable safety profile with an encouraging efficacy in SLE patients. These preliminary results support further evaluation in larger clinical trials to better define the benefit–risk of ATO in SLE and other systemic immune-mediated diseases.
Abbreviations
ACR:American College of Rheumatology; ANA: Anti-nuclear antibodies; ATO: Arsenic trioxide; APL: Acute promyelocytic leukaemia;
AZA: Azathioprine; BILAG: British Isles Lupus Assessment Group; cGVHD: Chronic graft versus host disease; DLT: Dose-limiting toxicity; HCQ: Hydroxychloroquine; IV: Intravenous; LLDAS: Lupus low disease activity state; MMF: Mycophenolate mofetil; MTX: Methotrexate; OCS: Oral corticosteroids; PGA: Physician’s global assessment; SELENA-SLEDAI: Safety of Estrogens in Lupus Erythematosus: National Assessment–SLE Disease Activity Index; SLE: Systemic lupus erythematosus; SFI: SELENA-SLEDAI Flare Index; SRI-4-R: SLE response index-4 response
Supplementary Information
The online version contains supplementary material available athttps://doi. org/10.1186/s13075-021-02454-6.
Additional file 1: Figure S1. LUPSENIC study scheme. A loading dose of ATO was administered intravenously each of the first 4 days (as in-patients), then twice a week (as outpatients) during weeks 2 to 4. Figure S2. Dose escalation scheme (Continual Reassessment Method): The ATO doses were studied sequentially; patients were planned to receive 0.1 mg/kg, 0.15 mg/kg or 0.20 mg/kg. If a patient experienced possible dose limiting toxicity (DLT), defined as grade 3 non reversible AE and/or grade 4 toxicity or death), a mathematical model was used to estimate the ob-served toxicity probability and determine the ATO dose for the next in-cluded patient. The maximum tolerated Dose (MTD) had been defined as the dose at which a DLT occurred in 10% of patients.
Acknowledgements
The authors thank the other investigators involved in the study: Emmanuel Châtelus, Yohann Foucher, Julie Graveleau, Jean-Robert Harlé, Micheline Pha, Mélanie Saint-Jean and Nicolas Schleinitz. The authors would also like to thank the patients, Sandrine Gardes, the staff of Medsenic (Catherine Lemon-nier, Dominique Gaida and Veronique Pomi-Schneiter), Anne-Marie Germond and Arnaud Laurent for active administrative support.
Authors’ contributions
MH and AN contributed to the study design, data analyses and
interpretation. EH, ZA, ME, JS, JFV and BG recruited patients and participated in data collection. JP performed the pharmacological analysis, JBH, the quality of life data analyses, and CV data analyses. FR was involved in the coordination of the trial. All authors provided final approval of the submitted manuscript.
Funding
MEDSENIC funded the study, but did not participate in the design of the study and collection, analysis and interpretation of data and in writing the manuscript. Nantes hospital (CHU Nantes), the academic institution of Mohamed Hamidou, Antoine Néel, Benjamin Gaborit, Christelle Volteau and Jean Benoit Hardouin, received grant for the current study from MEDSENIC, a commercial sponsor.
Availability of data and materials
The datasets analysed during the current study are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
The study complied with the Declaration of Helsinki for the conduct of research in humans and was approved by the Ethics Committee of Tours hospital, France. All patients provided written informed consent.
Consent for publication Not applicable
Competing interests
François Rieger is currently an employee of MEDSENIC. Author details
1Department of Internal Medicine, CHU Nantes, Nantes Université, Nantes,
France.2Department of Biological Toxicology, AP-HP, Lariboisière Hospital,
University Paris VII, Paris, France.3Department of Internal Medicine 2, Centre
National de Référence pour le Lupus, Institut E3M, Hôpital Pitié-Salpétrière, Paris, France.4Service de Médecine Interne, Aix Marseille Univ, APHM, CNRS,
INSERM, CIML, Hôpital de la Timone, Marseille, France.5Department of
Rheumatology, University of Strasbourg, Strasbourg, France.6Department of
Internal Medicine, Haut-Lévêque University Hospital, Bordeaux, France.
7Plateforme de Méthodologie et Biostatistiques, CHU Nantes, Université de
Nantes, Nantes, France.8INSERM UMR 1246-SPHERE, Université de Nantes,
Nantes, France.9Department of Internal Medicine, Centre de Référence des
Maladies Autoimmunes Systémiques Rares du Nord et Nord-Ouest de France (CeRAINO), University of Lille, Lille, France.10MEDSENIC, SAS, a company with
CNRS participation, Strasbourg, France.
Received: 17 April 2020 Accepted: 18 February 2021
References
1. Lo MS, Tsokos GC. Recent developments in systemic lupus erythematosus pathogenesis and applications for therapy. Curr Opin Rheumatol. 2018;30: 222–8.
2. Navarra SV, Guzmán RM, Gallacher AE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377:721–31. 3. Touma Z, Gladman DD. Current and future therapies for SLE: obstacles and
recommendations for the development of novel treatments. Lupus Sci Med. 2017;4:e000239.
4. Merrill JT, Neuwelt CM, Wallace DJ, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62:222–33.
5. Rovin BH, Furie R, Latinis K, Looney RJ, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 2012;64:1215–26. 6. Alexander T, Sarfert R, Klotsche J, et al. The proteasome inhibitor
bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann Rheum Dis. 2015;74:1474–8. 7. Mathews V, George B, Lakshmi KM, et al. Single-agent arsenic trioxide in the
treatment of newly diagnosed acute promyelocytic leukemia: durable remissions with minimal toxicity. Blood. 2006;107:2627–32.
8. Emadi A, Gore SD. Arsenic trioxide - an old drug rediscovered. Blood Rev. 2010;24:191–9.
9. Roboz GJ. Arsenic and old lace: novel approaches in elderly patients with acute myeloid leukemia. Semin Hematol. 2008;45(Suppl 2):S22–4. 10. Zhou J, Zhang Y, Li J, et al. Single-agent arsenic trioxide in the treatment of
children with newly diagnosed acute promyelocytic leukemia. Blood. 2010; 115:1697–702.
11. Mathews V, George B, Chendamarai E, et al. Single-agent arsenic trioxide in the treatment of newly diagnosed acute promyelocytic leukemia: long-term follow-up data. J Clin Oncol. 2010;28:3866–71.
12. Bobé P, Bonardelle D, Benihoud K, et al. Arsenic trioxide: a promising novel therapeutic agent for lymphoproliferative and autoimmune syndromes in MRL/lpr mice. Blood. 2006;108:3967–75.
13. Zhao Y, Wen G, Qiao Z, et al. Effects of tetra-arsenic tetra-sulfide on BXSB lupus-prone mice: a pilot study. Lupus. 2013;22:469–76.
14. Kavian N, Marut W, Servettaz A, et al. Reactive oxygen species-mediated killing of activated fibroblasts by arsenic trioxide ameliorates fibrosis in a murine model of systemic sclerosis. Arthritis Rheum. 2012;64:3430–40. 15. Kavian N, Marut W, Servettaz A, et al. Arsenic trioxide prevents murine sclerodermatous graft-versus-host disease. J Immunol. 2012;188:5142–9. 16. Hu H, Chen E, Li Y, et al. Effects of arsenic trioxide on INF-gamma gene
expression in MRL/lpr mice and human lupus. Biol Trace Elem Res. 2018;184: 391–7.
17. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
18. Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25: 1271–7.
19. Gladman DD, Ibañez D, Urowitz MB. Systemic lupus erythematosus disease activity index 2000. J Rheumatol. 2002;29:288–91.
20. Raffoux E, Rousselot P, Poupon J, et al. Combined treatment with arsenic trioxide and all-trans-retinoic acid in patients with relapsed acute promyelocytic leukemia. J Clin Oncol. 2003;21:2326–34.
21. O’Quigley J, Pepe M, Fisher L. Continual reassessment method: a practical design for phase 1 clinical trials in cancer. Biometrics. 1990;46:33–48. 22. O’Quigley J, Shen LZ. Continual reassessment method: a likelihood
approach. Biometrics. 1996;52:673–84.
23. Isenberg DA, Rahman A, Allen E, et al. BILAG 2004. Development and initial validation of an updated version of the British Isles Lupus Assessment Group’s disease activity index for patients with systemic lupus erythematosus. Rheumatology. 2005;44:902–6.
24. Petri M, Kim MY, Kalunian KC, et al. Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med. 2005;353:2550–8. 25. Buyon JP, Petri MA, Kim MY, et al. The effect of combined estrogen and
progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: a randomized trial. Ann Intern Med. 2005;142:953–62. 26. Furie RA, Petri MA, Wallace DJ, et al. Novel evidence-based systemic lupus
erythematosus responder index. Arthritis Rheum. 2009;61:1143–51. 27. Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for
acute promyelocytic leukemia. N Engl J Med. 2013;369:111–21. 28. Franklyn K, Lau CS, Navarra SV, et al. Definition and initial validation of a
Lupus Low Disease Activity State (LLDAS). Ann Rheum Dis. 2016;75:1615–21. 29. Morand EF, Trasieva T, Berglind A, et al. Lupus Low Disease Activity State
(LLDAS) attainment discriminates responders in a systemic lupus erythematosus trial: post-hoc analysis of the Phase IIb MUSE trial of anifrolumab. Ann Rheum Dis. 2018;77:706–13.
30. Ordi-Ros J, Sáez-Comet L, Pérez-Conesa M, et al. Enteric-coated
mycophenolate sodium versus azathioprine in patients with active systemic lupus erythematosus: a randomised clinical trial. Ann Rheum Dis. 2017;76: 1575–82.
31. Zen M, Iaccarino L, Gatto M, et al. The effect of different durations of remission on damage accrual: results from a prospective monocentric cohort of Caucasian patients. Ann Rheum Dis. 2017;76:562–5. 32. Bernatsky S, Ramsey-Goldman R, et al. Smoking is the most significant
modifiable lung cancer risk factor in systemic lupus erythematosus. J Rheumatol. 2018;45:393–6.
33. Zhu H, Hu J, Chen L, et al. The 12-year follow-up of survival, chronic adverse effects, and retention of arsenic in patients with acute promyelocytic leukemia. Blood. 2016;128:1525–8.
34. Xu W, Li X, Quan L, et al. Arsenic trioxide decreases the amount and inhibits the function of regulatory T cells, which may contribute to its efficacy in the treatment of acute promyelocytic leukemia. Leuk Lymphoma. 2018;59:650–9. 35. Zhao B, Xia J-J, Wang L-M, et al. Immunosuppressive effect of arsenic
trioxide on islet xenotransplantation prolongs xenograft survival in mice. Cell Death Dis. 2018;9:408.
36. Xu S, Chen J, Wang F, et al. Arsenic trioxide combined with co-stimulatory molecule blockade prolongs survival of cardiac allografts in alloantigen-primed mice. Transpl Immunol. 2010;24:57–63.
37. Macoch M, Morzadec C, Fardel O, et al. Inorganic arsenic impairs differentiation and functions of human dendritic cells. Toxicol Appl Pharmacol. 2013;266:204–13.
38. Ye Y, Ricard L, Siblany L, Stocker N, De Vassoigne F, Brissot E, Lamarthée B, Mekinian A, Mohty M, Gaugler B, Malard F. Arsenic trioxide induces regulatory functions of plasmacytoid dendritic cells through interferon-α inhibition. Acta Pharm Sin B. 2020;10:1061–72.
39. Wang G, Pierangeli SS, Papalardo E, et al. Markers of oxidative and nitrosative stress in systemic lupus erythematosus: correlation with disease activity. Arthritis Rheum. 2010;62:2064–72.
40. Li C, Zhang J, Wang W, Wang H, Zhang Y, Zhang Z. Arsenic trioxide improves Treg and Th17 balance by modulating STAT3 in treatment-naive rheumatoid arthritis patients. Int Immunopharmacol. 2019;73:539–51. 41. Robert M, Miossec P. Interleukin-17 and lupus: enough to be a target ? For
which patients ? Lupus. 2020;29:6–14.
42. Torka P, Al Ustwani O, Wetzler M, et al. Swallowing a bitter pill-oral arsenic trioxide for acute promyelocytic leukemia. Blood Rev. 2016;30:201–11. 43. Au W-Y, Kumana CR, Lee HKK, et al. Oral arsenic trioxide-based maintenance
regimens for first complete remission of acute promyelocytic leukemia: a 10-year follow-up study. Blood. 2011;118:6535–43.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.

![[Hepatitis E: from zoonotic transmission to chronic infection in immunosuppressed patients.]](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)