i
TABLE DES MATIERES
Introduction générale
……… 1Chapitre I : Connaissances de base de la spectroscopie
I.1 Historique ... 2I.2 Le spectre électromagnétique………... 3
I.3 Energie de molécule………... 3
I.4 Population des niveaux d’énergie………... 4
I.5 Interaction des ondes électromagnétiques avec la matière ……… 5
I.5.1 Absorption……….. 5
I.5.2 Emission………. 6
I.1.3 Diffusion ………... 6
I.6 Loi de Beer- Lambert ………... 7
I.7 Les types de spectres……… I.7.1 Spectres de raies………... I.7.2 Spectres de bandes……… I.8 Méthodes spectroscopiques……… I.8.1 Spectroscopie vibrationnelle………... I.8.2 Spectroscopie UV-Visible……….. I.8.3 Spectroscopie RMN………. I.8.4 Spectroscopie résonance paramagnétique électronique (RPE)………
Chapitre II : Spectroscopie infrarouge
8 8 8 9 10 13 13 14 II. 1 Introduction………... 15II. 1 Rotation des molécules
………..
16II.2.1 Rotateur rigide ………...
II.2.1.1Règles de sélection………
16 19 II.2.2 Rotateur non rigide ……….. 20ii
II.3.1 Molécule diatomique………...
II.3.1.1 Oscillateur harmonique………...22 22 II.3.1.2. Les règles de sélection………
II.3.1.3 Oscillateur anharmonique………...
II.3.2 Molécules polyatomiques………. II.3.2.1 Les modes de vibration……….. II.3.2.2 Règle de sélection………... II.3.2.3 Dénombrement des vibrations………28 30 34 36 37 39
II.4
Couplage rotation- vibration………... 43
Chapitre III Techniques expérimentales en spectroscopie infrarouge
III.1 Introduction……….. 45III.2 Spectromètre dispersif………. 45
III.2.1 Les éléments constituant un spectromètre IR dispersif………. 46
III.2.1.1 Sources lumineuses de radiations IR………. 46
III.2.1.2 Monochromateur... 46
III.2.1.3 Détecteur……… 46
III.3 Spectromètre non dispersif……….
III.3.1 Interféromètre de Michelson………...
III.3.2 Fonctionnement……… III.3.3 Avantages……… 47 47 47 50 III.4 Résolution………...……….………... 51III.5 Préparation de l’échantillon………..………... 51
Chapitre IV Partie expérimentale
IV.1 Introduction………..… 52IV.2 Mousse de polyuréthane………..……….... 52
IV.3 Préparation de la mousse polyuréthane ……… 53
IV.4 Charbon actif ………. 54
IV.5 Utilisation de la mousse polyuréthane ……… 54
iii
Chapitre V : Analyse et résultats
V.1 Introduction………..………. V.2 Interprétation des spectres………..
56 56
V.3 Caractérisation des échantillons ……….……… 57
V.3.1 Caractérisation de la mousse PU ……….…….. V.3.2 Caractérisation de la mousse PUCAG10………
57 61
Conclusion générale
……… 64Bibliographie
………. 65Introduction
Page 1
La spectroscopie est l’analyse du rayonnement électromagnétique émis, absorbé ou diffusé par la matière, par lesquelles des molécules effectuent une transition d’un état quantique à un autre. Elle permet d’expliquer des phénomènes qui nous entourent, elle sera un outil fondamental pour l’identification des substances inconnues (liquide, solide ou gazeuse) du fait de l’interaction des rayonnements électromagnétiques avec la matière.
La spectroscopie a des applications dans de nombreux domaines : astrophysique, chimie, biologie, pharmacie…etc. Grâce à la spectroscopie, on a pu développer les lasers servant à lire les codes-barres dans les grandes surfaces, permettant le fonctionnement des imprimantes laser. Dans les laboratoires de recherche, la spectroscopie offre d’innombrables possibilités d’observations et de compréhension des phénomènes chimiques.
A chacun des domaines particuliers du rayonnement électromagnétique correspond un type de spectroscopie qui repose sur une interaction particulière de la matière avec ce rayonnement. Ces types de spectroscopie sont des techniques complémentaires.
Dans notre mémoire nous allons voir comment il est possible d’interpréter les résultats de l’interaction pour déduire des informations quant à la structure atomique et moléculaire irradiée par un rayonnement infrarouge. Le type d’information obtenu dépendra de la sensibilité de l’appareillage, la méthode d’enregistrement du spectre ainsi que la nature d’échantillon (liquide, solide ou gazeuse).
Dans notre travail, nous avons essayé de caractériser à partir des spectres de deux échantillons qui ont été enregistrés au LCME (Laboratoire de Chimie Moléculaire et Environnement), de l’Université de Savoie, France en collaboration avec le Laboratoire SEA2M (Structure et Application des Matériaux Moléculaires) FSET, Université de Mostaganem, sous la direction de M. N. Ben derdouche. Les échantillons sont :
La mousse polyuréthane brut (PU).
La mousse polyuréthane imprégné avec le charbon actif commercial (PUCAG10). Dans les deux premiers chapitres, nous avons rappelé les notions générales de la spectroscopie en développant beaucoup plus la méthode de spectroscopie infrarouge. Dans le troisième chapitre on a décrit deux techniques expérimentales : spectromètre à double faisceaux, spectromètre à transformée de Fourier. Dans le quatrième chapitre, est la partie expérimentale, on a défini la mousse polyuréthane, et la méthode de préparation de la mousse. Au dernier chapitre, nous avons essayé d’exploiter les résultats, en analysant chaque transition vibrationnelle dans les deux spectres.
Introduction
Page 2
iv
Chapitre I Connaissances de base de la spectroscopie
Figure I.1 Expérience de d’Isaac Newton. ………... 2
Figure I.2 Spectre électromagnétique………... 3
Figure I.3 Diagramme énergétique……… .……… 4
Figure I.4 Schéma illustré le processus d’absorption.……… 5
Figure I.5 Schéma illustré le processus d’émission……… 6
Figure I.6 Schéma illustré le processus de diffusion……….. 6
Figure I.7 Schéma illustrant la loi de Beer- Lambert………... 7
Figure I.8 Variation de l’intensité I en fonction de l’épaisseur de la cuve... 7
Figure I.9 Spectre de raie ……… 8
Figure I.10 Spectre de bande ……….….. 9
Figure I.11 Mécanismes de diffusion Raman et Rayleigh……….……….. 10
Figure I.12 Représentation schématique d’un spectre Raman……..……….. 11
Figure I.13 Spectre Raman de CCl4 excité par une radiation laser avec λ= 488 nm.. 12
Figure I.14 Vibrations de molécule ……….. 12
Chapitre II Spectroscopie infrarouge Figure II.1 Domaines de l’IR dans le spectre électromagnétique ……… 14
Figure II.2 Représentation schématique d’une molécule diatomique hétéronucléaire. 15 Figure II.3 Modèle du rotateur rigide équivalent à une molécule diatomique ……… 16
Figure II.4 Représentation des niveaux d’énergie d’un rotateur rigide ………... 19
Figure II.5 Modèle simple de l’oscillateur harmonique dans le cas d’une molécule diatomique hétéronucléaire ………. 21
v
Figure II.7 Influence de la force de liaison et de la masse atomique sur la fréquence. 23
Figure II.8 Courbe de potentiel de l'oscillateur harmonique quantique ……….. 26
Figure II.9 Représentation schématique d’une molécule diatomique hétéronucléaire équivalente à un dipôle électrique simple……….. 27
Figure II.10 Différence entre la courbe de potentiel de l'oscillateur harmonique et de l’oscillateur anharmonique ……… 29
Figure II.11 Courbe de potentielle de Morse ……….……….. 31
Figure II.12 Nomenclateur des bandes de vibration ……… 33
Figure II.13 Vibration d’élongation de la molécule d’eau ………... 40
Figure II.14 Représentation schématique d’une bande de rotation-vibration d’une molécule diatomique ………... 43
Chapitre III Techniques expérimentales en spectroscopie infrarouge Figure III.1 Schéma de principe d’un spectromètre IR dispersif à double faisceaux... 45 Figure III.2 Composants d’un monochromateur………... 46
Figure III.3 Schéma de l’interféromètre de Michelson ……….. 47
Figure III.4 Spectre et son interférogramme correspond………. 49
Figure III.5 Schéma d’un spectromètre de transformée de Fourier………. 50
Figure III.6 Deux bandes spectrales espacées de d et leur interférogramme ……….. 51
Chapitre IV Partie expérimentale Figure IV.1 Image obtenue par MEB (Microscopie Electronique à Balayage) de la mousse de polyuréthane………. Figure IV.2 Spectre IRTF de la mousse polyuréthane brut (PU)………... 53 55 Figure IV.3 Spectre IRTF de la mousse polyuréthane imprégnée par le charbon actif (PUCAG10)………... 55
Chapitre V Analyse et résultats Figure V.1 Différentes formes des bandes……….. 57
vi
Figure V.2 Spectre IRTF de la mousse polyuréthane brut (PU)……… 57 Figure V.3 Spectre infrarouge de la réaction isocyanate - alcool. Variation de
l’absorbance du pic d’isocyanate N=C=O au cours du temps……….. 60
Figure V.4 Spectre IRTF de la mousse polyuréthane imprégnée par le charbon actif
(PUCAG10)……….. 61
Figure V.5 Les spectres IR (CAG10, PU et PUCAG10)……….. 63
iv
Chapitre I Connaissances de base de la spectroscopie
Tableau I.1 Les processus d’excitation correspondent à chaque Domaine
de fréquences ……… 9
Chapitre II Spectroscopie infrarouge
Tableau II.1 Spectre rotationnel partiel de HCl………...…. 21
Tableau II.2 Les constantes de quelques molécules diatomiques 32
Tableau II.3 Table de caractère de groupe ……….. 39
Chapitre V Analyse et résultats
Tableau V.1 : Attributions des bandes de spectre infrarouge de PU……….. Tableau V.2 : Attributions des bandes de spectre infrarouge de PUCAG10..
58 62
Chapitre I : Connaissances de base de la spectroscopie
Page 2
I.1 Historique :
Le point de départ de la spectroscopie est constitué par les travaux d’Isaac Newton. Il explique notamment que la lumière blanche de soleil est composée des rayons lumineux correspondant à des couleurs différentes lors de leur passage a travers un prisme [1]. Ces couleurs sont en fait une succession de radiations visibles de longueurs d’onde continuellement variables. Newton montra ensuite que les couleurs du spectre ne peuvent pas se décomposer en de nouvelles couleurs : si on envoie de la lumière verte sur un prisme, on retrouve la même lumière en sortie. Cette lumière est dite monochromatique. Premier prisme Seconde prisme Lumière visible
Figure I.1: Expérience de d’Isaac Newton.
En 1800, William Herschel fit l’expérience avec un thermomètre de mesurer la température dans différentes zones du spectre solaire; il montra qu’une chaleur existait au delà du rouge dans le domaine spectral. Il appela ces radiations les infrarouges [2]. En 1801, Ritter démontra que des radiations existaient au delà du violet.
En 1802 W.H. Wollaston observe quelques raies sombres dans le spectre solaire. Des observations plus précises réalisées par J. Fraunhofer en 1814, permettent de dénombrer un nombre beaucoup plus élevé des raies sombres dans ce même spectre solaire, il définit ces raies par des lettres de l’alphabet (raie D du sodium, raies H et K du calcium ionisé.)
Après les travaux de Fraunhofer, et en 1859 le chimiste Bunsen et le physicien Kirchhoff reposent sur l’analyse des spectres des flammes pour détecter les éléments chimiques. Ces
observations conduisent W.H.F. Talbot à formuler en 1862 le principe de l'analyse chimique basé sur l'observation du spectre [1].
Chapitre I : Connaissances de base de la spectroscopie
Page 3
I.2 Le spectre électromagnétique :
Le rayonnement électromagnétique est une forme d’énergie constituée d’ondes caractérisés par : Une vitesse de propagation c = 3.108 m/s, elle est constante pour toutes les ondes
électromagnétiques dans le vide.
Une fréquence 𝜈 exprimée en hertz (Hz) ou un nombre d’onde 𝜈̅ (cm−1). Une longueur d’onde λ (nm).
Ces trois grandeurs sont liées par la relation : λ =c
𝜈 = 1
𝜈̅ (I. 1) Le rayonnement électromagnétique divisé en plusieurs régions depuis les ondes hertziennes basses d'énergie jusqu'à le domaine des rayons γ et des rayons hauts d’énergie [3].
Énergie Nombre 109 106 2.5× 104 1.25× 104 10 0.03 (cm−1) d’onde (𝜈̅) Fréquence 3 × 1019 3 × 1016 7.5 × 1014 3.75 × 1014 3 × 1011 109 (Hz) (𝜈) Longueur 0.01 nm 10 nm 400 nm 800 nm 1000 µm 30 cm d’onde (λ)
Figure I.2 : Spectre électromagnétique.
I.3 Energie de molécule :
La molécule possède un mouvement interne des électrons, un mouvement de rotation et un
mouvement de vibration des atomes. Tous ces mouvements sont quantifiés. On remarque que les mouvements de translation ne sont pas quantifiés on ne les prend pas en compte.
L’énergie d’une molécule en première approximation (approximation de Born-Oppenheimer) est écrite sous la forme suivante [1] :
E = Eel+ Evib+Erot (I. 2) Eel : Energie électronique (Mouvement des électrons).
Evib : Energie vibrationnelle (Oscillation de noyaux).
Erot : Energie rotationnelle (Rotation d’ensemble de la molécule). Ces trois énergies ont des ordres de grandeurs très différentes :
Eel ≫ Evib ≫ Erot (I. 3)
Rayon γ Rayon x Ultraviolet Visible Infrarouge Microonde Ondes radio
Chapitre I : Connaissances de base de la spectroscopie
Page 4
Les niveaux d’énergie électronique, vibrationnelle et rotationnelle sont représentés par un diagramme énergétique et par des nombres quantiques n, v et J.
n : Nombre quantique électronique.
v : Nombre quantique vibrationnelle. J : Nombre quantique rotationnelle.
v = 1 J = 2 J = 1
État électronique excité v = 2 J = 2 Niveau vibrationnelle J = 1 v = 1 J = 2 Niveau rotationnelle J = 1
État électronique fondamental
Figure I.3 : Diagramme énergétique.
I.4 Population des niveaux d’énergie :
Le nombre des particules sur un niveau énergétique i donné s’appelle la population notée Ni. La population sur chaque niveau par rapport à la population du niveau fondamental N0 obéit à la loi de distribution de Maxwell - Boltzmann :
Ni N0 = ( gi g0) e −(Ei−E0) KBT (I. 4)
Ni: Nombre de particules (atome, ion ou molécule) sur l'état excité i. N0: Nombre de particules sur l'état fondamental 0.
gi , g0 : Facteur de dégénérescence des états i et 0 respectivement.
gi = 1 ⟹ Pour la vibration.
gi = (2J+1) ⟹ Pour la rotation.
Chapitre I : Connaissances de base de la spectroscopie
Page 5
Ei, E0: Energie des états i et 0 respectivement. k : Constante de Boltzmann (=1,38.10-23 J.K−1). T : Température en Kelvin(K).
En utilisant cette relation, on montre que dans la plupart des cas, à la température ordinaire : Plusieurs niveaux de rotation sont largement peuplés.
Les niveaux excités de vibration sont peu peuplés.
Toutes les molécules sont dans l’état fondamental électronique.
I.5 Interaction des ondes électromagnétiques avec la matière [4] :
Selon Planck, les échanges d'énergie entre matière et rayonnement se réalisent par quanta d'énergie :
∆E = h 𝜈 (I. 5) ∆E : Différence d’énergie exprimée en joule (J).
h: Constante de Planck (= 6,624.10−34 J.s).
Trois processus sont à la base de tous les phénomènes spectroscopiques (Absorption, émission et diffusion).
I.5.1 Absorption :
Si un système matériel est soumis à l’action d’un faisceau de lumière d’énergie donnée, un photon peut être absorbé. Le système passe du niveau d’énergie Ei au niveau d’énergie Ef.
Avec : h𝜈 = Ef− Ei Ef > Ei (I. 6)
Ef h𝜈if
Ei
Figure I.4 : Schéma illustré le processus d’absorption.
Les états Ei et Ef sont caractéristiques d’un niveau rotationnel, vibrationnel et électronique
Pour obtenir une transition purement rotationnelle, seul le nombre quantique de rotation varie. L’énergie d’excitation se situe dans les microondes ou le lointain infrarouge. Le nombre quantique de vibration v varie, nous avons une transition vibrationnelle. Sa
fréquence se situe dans l’infrarouge.
Le nombre quantique électronique n peut aussi varier, nous avons alors une transition électronique. Sa fréquence se situe dans le visible ou l’ultraviolet.
Chapitre I : Connaissances de base de la spectroscopie
Page 6
I.5.2 Emission :
Un système d’énergie
Ef peut émettre spontanément un photon pour descendre sur un niveau inférieur E i tel que :∆E = hν = Ef− Ei Ef
hʋif
Ei
Figure I.5 : Schéma illustré le processus d’émission.
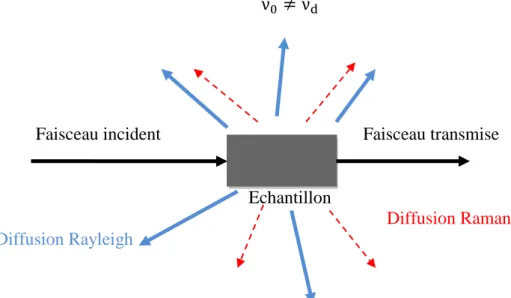
I.5.3 Diffusion [2] :
Le choc entre la matière et une radiation de fréquence ν0 peut renvoyer le photon dans une autre direction, avec ou sans modification de son énergie. On dit qu’il y a diffusion.
Lorsque l’énergie des ondes diffusées reste inchangée par rapport à l’énergie des ondes excitatrices, le choc est dit élastique. Ceci correspond à la diffusion Rayleigh ou diffusion élastique, qui conserve la fréquence de l’onde incidente.
ν0= 𝜈𝑑 νd: Fréquence de l’onde diffusée.
Lorsque l’énergie des ondes diffusées change par rapport à l’énergie des ondes excitatrices, le choc est dit inélastique. Ce phénomène porte le nom de diffusion Raman ou diffusion inélastique.
ν0 ≠ νd
Faisceau incident Faisceau transmise
Echantillon
Diffusion Raman
Diffusion Rayleigh
Chapitre I : Connaissances de base de la spectroscopie
Page 7
I.6 Loi de Beer- Lambert [4; 5] :
Soit un faisceau parallèle de lumière monochromatique d’intensité 𝐈𝟎 traversant une solution absorbante de concentration c dans une cuve d’épaisseur 𝒍. Une partie de ce rayonnement sera absorbée par l’échantillon et une partie sera transmise 𝐈.
Echantillon
𝐈
𝟎𝐈
𝒍
Figure I.7: Schéma illustrant la loi de Beer- Lambert.
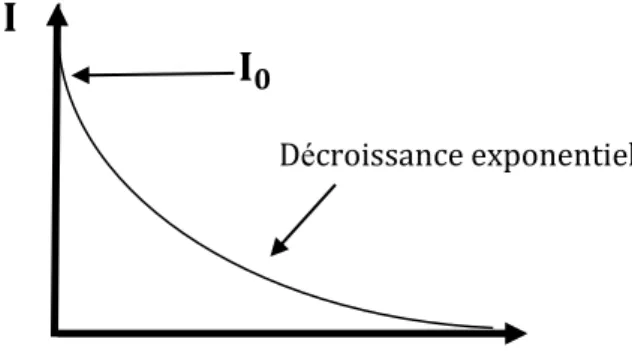
L’intensité de la lumière transmise I décroit de façon exponentielle en fonction de l’épaisseur l selon la formule suivante :
I = I0 e−ε𝑙c (I. 7)
I0 : Intensité de la radiation incidente.
I : Intensité de la radiation transmise. c : Concentration molaire (mol.l−1). 𝑙 : Épaisseur de la cuve (cm).
ε : Coefficient d’extinction (mol.l−1cm−1).
𝐈
𝐈
𝟎Décroissance exponentielle 𝒍
Figure I.8 : Variation de l’intensité I en fonction de l’épaisseur de la cuve.
La loi de Beer-Lambert est exprimée sous la forme suivante : logI0
Chapitre I : Connaissances de base de la spectroscopie
Page 8
Le coefficient d’extinction ε est une constante caractéristique de l’échantillon étudiée à une longueur d'onde donnée λ et l’épaisseur de la cuve 𝑙 donnée.
On définit :
A = logI0
I (I. 9) T = I
I0 (I. 10) A : Absorbance (Sans unité).
T : Transmission (Sans unité). On obtient alors la relation :
A = − log T = ε𝑙c (I. 11)
I.7 Les types de spectres
[2] :L’interaction de la matière avec un rayonnement électromagnétique provoque des transitions qui se manifestent par des spectres de raies ou de bandes.
I.7.1 Spectres de raies:
Dans un atome, une variation de l’énergie électronique donne naissance à une seule raie spectrale. La position de chaque raie correspond à une radiation monochromatique.
E Intensité Etat excité Excitation Etat fondamental 𝜆
Figure I.9: Spectre de raie.
I.7.2 Spectres de bandes
Théoriquement, le spectre d’une molécule est un spectre de raies (quantification, valeurs discrètes d’énergie). Cependant, outre les niveaux électroniques associés aux électrons des atomes qui les constituent, il existe d’autres niveaux d’énergie correspondant à leurs énergies de rotation et vibration entre lesquels on peut expérimentalement observer des transitions.
Ainsi, une transition entre deux niveaux électroniques peut conduire à une modification des énergies à la fois de vibration et de rotation, donc à un ensemble de transitions d’énergies très voisines ce qui conduit à un spectre de bandes.
Chapitre I : Connaissances de base de la spectroscopie
Page 9
E Intensité Etat excité
Etat fondamental λ
Figure I.10: Spectre de bande.
I.8 Méthodes spectroscopiques:
A chacun des domaines particuliers du rayonnement électromagnétique correspond un type de spectroscopie qui repose sur une interaction particulière de la matière avec ce rayonnement.
Domaines de fréquence Processus d’excitation
Rayons γ et x Electrons des orbitales atomiques de cœur
UV et visible Electrons de l’orbital
atomique périphérique Infrarouge (IR) Vibration des liaisons
ou rotation
Micro-ondes Rotations
Ondes radio Spins nucléaires
Tableau I.1 : Les processus d’excitation correspondent à chaque Domaine de fréquences [6].
Les méthodes spectroscopiques les plus utilisées sont la spectroscopie vibrationnelle, spectrophotométrie ultraviolet-visible, spectroscopie de résonance magnétique nucléaire (RMN) et spectroscopie de résonance paramagnétique électronique (RPE).
Chapitre I : Connaissances de base de la spectroscopie
Page 10
I.8.1 Spectroscopie vibrationnelle :
La spectroscopie vibrationnelle est le terme utilisé pour décrire deux techniques d'analyse
spectroscopie infrarouge et Raman. Les deux spectroscopies sont des outils non destructifs, fournissent des informations sur la composition moléculaire et la structure. Ces techniques mesurent les niveaux d’énergie vibrationnelle qui sont associés à des liaisons chimiques dans l'échantillon.En spectroscopie infrarouge, l'échantillon est irradié avec une lumière polychromatique et un photon de la lumière est absorbé lorsque la fréquence (énergie) de la lumière absorbée correspond à l'énergie requise pour une liaison particulière à vibrer dans l'échantillon. Pour un mouvement de vibration actif en infrarouge, il faut accompagner d’une variation de moment dipolaire de la molécule. Les molécules diatomiques symétriques N2, O2… ont un moment dipolaire nul, elles n’absorbent pas l’infrarouge.
L’intensité des raies dépend de trois facteurs : La probabilité de transition.
La population des niveaux de départ. La quantité de matière (concentration).
L’étude de probabilité de transition conduit à établir des règles de sélection. Si deux transitions sont caractérisées par des probabilités de transition égales, le plus intense sera celle émis par le niveau dont la population est plus élevée, généralement, le rapport des populations de deux niveaux est obtenu à partir de la loi de Boltzmann [1; 2].
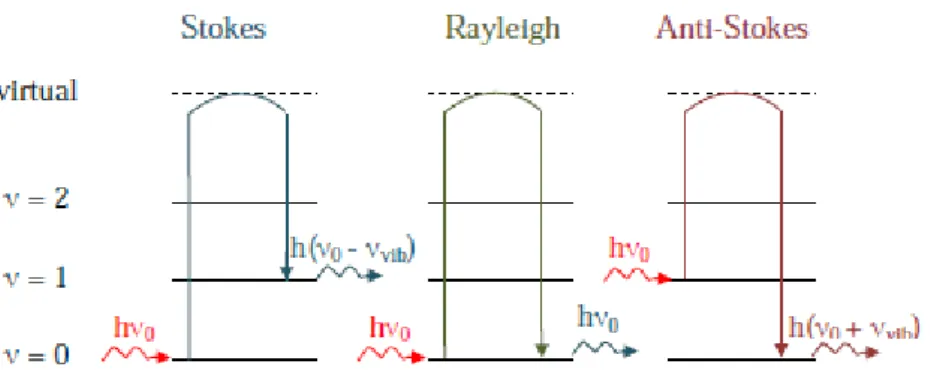
En spectroscopie Raman, l'échantillon est irradié avec une lumière monochromatique de type laser de fréquence ʋ0. Elle réémet ensuite une radiation, cette radiation comporte deux types de signaux. Le premier correspond à la diffusion Rayleigh, la radiation incidente est diffusée élastiquement sans changement d’énergie. Le second type connu sous le nom diffusion Raman, la radiation incidente diffusée inélastiquement [7].
Si la fréquence de radiation diffusée ʋ1 inférieur à la fréquence de radiation incidente ʋ0, (ʋ1= ʋ0− ʋVib). Il y a gain d’énergie vibrationnelle pour la molécule et on parle des raies stokes [8].
Si la fréquence de radiation diffusée 𝜐2 supérieur à la fréquence de radiation incidente ʋ0 , ( ʋ2 = ʋ0+ ʋVib). Il y a perte d’énergie vibrationnelle pour la molécule, ce qui correspond aux raies anti-Stokes [8].
Chapitre I : Connaissances de base de la spectroscopie
Page 11
Figure I.11: Mécanismes de diffusion Raman et Rayleigh.
Pour qu’une vibration soit active en Raman lorsque la molécule vibre, il doit y avoir une variation du moment dipolaire induit.
P⃗⃗ = α E ⃗⃗⃗ (I. 12) P⃗⃗ : Moment dipolaire induit.
α : Polarisabilité. E⃗⃗ : Champ électrique.
Autrement dit, une modification de la polarisabilité 𝛼, le nuage électronique de la molécule peut se déformer (changement dans la forme, la taille ou l'orientation) sous l’effet d’un champ électrique appliqué [9].
𝜕𝛼
𝜕𝑄 ≠ 0 (I. 13) Q : Représente les coordonnées normales et décrit les déplacements au cours de la vibration. La représentation des spectres Raman fait en fonction du déplacement Raman 𝜹 [𝟏𝟎] :
δ (cm−1) = (1 λi−
1
λr) (I. 14) λr ∶ Longueur d’onde de la raie Raman.
λi : Longueur d’onde de la lumière incidente.
Chapitre I : Connaissances de base de la spectroscopie
Page 12
L’intensité d’une raie de diffusion Raman dépend du nombre de molécules présentes dans l’état initial. Comme le montre la figure I.11, l’excitation dans le cas de la diffusion anti-Stokes se fait à partir d’un niveau énergétique supérieur à celui de la diffusion Stokes.
La probabilité pour que le système soit initialement dans son état fondamental ou dans un état vibrationnel excité est donnée par la distribution de Boltzmann (la probabilité d’avoir des molécules dans un état vibrationnel excité est plus faible que celle de les avoir dans un état stable). Le rapport d’intensité des raies anti-Stokes IAS sur Stokes IS est proportionnel au facteur de Boltzmann.
IAS IS ≈
Nv N0 ≈ e
−(Ev−E0) KT⁄ < 1 (I. 15)
Donc les bandes Stokes sont plus intenses que les bandes anti-Stokes [10].
Figure I.13 : Spectre Raman de CCl4 excité par une radiation laser avec λ= 488 nm
Enfin, le spectroscopie infrarouge et Raman sont des techniques complémentaires. Généralement, la spectroscopie IR utilisé pour une mesure des vibrations asymétriques de groupe non polaire, et la spectroscopie Raman convenable pour les vibrations symétriques de groupe non polaire [11].
− + −
(a) Active en Raman
(b) Active en IR
(c) Active en IR
Figure I.14 : Vibrations de molécule CO2,(a) mode d’élongation symétrique (b) mode d’élongation asymétrique
Chapitre I : Connaissances de base de la spectroscopie
Page 13
I.8.2 Spectroscopie UV-Visible [1] :
La spectroscopie UV-Visible est une technique d’analyse qualitative et surtout quantitative
grâce à l’utilisation de la loi de Beer-Lambert d’un grand nombre d’espèces inorganique et organique. Elle est basée sur l’absorption moléculaire dans l’ultraviolet et le visible.Les domaines de longueurs d’onde considérés ici sont :
Pour la radiation ultraviolette (UV) : 200-400 nm [50000-25000cm−1] le proche UV. pour la radiation du domaine visible 400-800 nm [25000-112500cm−1].
Ces radiations provoquent des transitions électroniques au sien de molécule
La spectroscopie UV-Visible est très utilisée pour étudier les structures électroniques des molécules, elle donne des informations globales sur la molécule, en particulier sur l'état de conjugaison de celle-ci, fort utile en chimie organique.
I.8.3 Spectroscopie RMN :
La spectroscopie résonance magnétique nucléaire est une technique non-destructive utilisée pour l’analyse des structures de nombreuses molécules chimiques. Elle sert principalement à la détermination structurale des composée organiques. Les principaux noyaux étudiés sont le proton H1 , le carbone 13C et le phosphore31P.
Cette technique repose sur le magnétisme nucléaire, les noyaux de certains atomes (1H, 13C, …) possèdent un moment magnétique nucléaire (sont considérés comme des aimants microscopiques caractérisées par une grandeur quantique spin).
La méthode de spectroscopie RMN est fondée sur la mesure de l’absorption d’une radiation dans le domaine des fréquences radio par un noyau atomique dans un champ magnétique fort. L’absorption d’une radiation électromagnétique peut provoquer une transition de spin, le noyau passant alors du niveau d’énergie le plus faible vers le niveau d’énergie le plus élevé. On dit qu’il y a résonance [5; 12].
Aujourd'hui, les applications de la RMN sont infinies et concernent de nombreux domaines de la physique, la chimie, la biologie et également l'imagerie médicale (l'imagerie par résonance magnétique ou IRM). [13]
Chapitre I : Connaissances de base de la spectroscopie
Page 14
I.8.4 Spectroscopie résonance paramagnétique électronique (RPE) :
La spectroscopie résonance paramagnétique électronique ou résonance de spin électrique détecte les espèces paramagnétiques.Le principe de RPE est basée sur l’absorption d’une radiation micro-onde par des électrons non-apparies (célibataire), lorsqu’ils sont placés dans un champ magnétique.
La spectroscopie résonance paramagnétique permet de mettre en évidence le caractère paramagnétique de l’échantillon, et obtient lesinformations sur la géométrie électronique et la structure. Cela concerne la chimie bio-inorganique, la biologie et la médecine.
Chapitre II : Spectroscopie infrarouge
Page 15
II.1 Introduction
La spectroscopie infrarouge est une classe de spectroscopie moléculaire,permet d’identifier la nature de liaisons chimiques présentes dans les molécules organiques et de déterminer les groupes fonctionnels. Elle est considérée comme un moyen de diagnostic non destructif (l’échantillon peut être récupéré).
La spectroscopie infrarouge repose sur l’absorption du rayonnement électromagnétique par l’échantillon, il est possible de déterminer la partie du rayonnement que l’échantillon absorbe en mesurant ce qui à été transmis. Cette radiation électromagnétique est découverte en 1800 par Frédéric Wilhelm Herschel, le domaine infrarouge s’étend de 0,8 à 1000 µm (10 à 12500 cm−1). Il est situé entre la région du spectre visible et micro-onde, peut être divisé en trois catégories selon les longueurs d’onde :
Le proche infrarouge entre 0,8 et 2,5 µm (4000 - 12500 cm−1). Le moyen infrarouge entre 2,5 et 25 µm (400 - 4000 cm−1). Le lointain infrarouge entre 25 et 1000 µm (10 - 400 cm−1).
Figure II.1 : Domaines de l’IR dans le spectre électromagnétique.
Suivant ces régions, des phénomènes différents sont observés en spectroscopie IR [9] :
En proche infrarouge le plus énergétique peut provoquer les vibrations complexes (harmoniques, combinaisons);
Le moyen infrarouge peut être utilisé pour étudier les vibrations et les rotations des molécules, cette région la plus riche en informations sur les structures des composés examinés ;
L’infrarouge lointain, faible énergie, peut être utilisé pour étudier les rotations des molécules.
Le rayonnement IR émis par la source polychromatique n’est généralement pas assez énergétique pour provoquer des transitions électroniques, mais il induit des transitions entre les niveaux d’énergie vibrationnelle et rotationnelle [1].
Chapitre II : Spectroscopie infrarouge
Page 16
II.2 Rotation des molécules :
La rotation est observée dans l’infrarouge pour les molécules dans l’état gazeux et
faiblement dans l’état liquide, par contre elle n’existe pas dans l’état solide.II.2.1 Rotateur rigide :
Le rayonnement infrarouge excite les mouvements de rotation des molécules. La mécanique
classique permet de calculer l’énergie de rotation d’une molécule diatomique 𝐄𝐫𝐨𝐭. L'énergie cinétique d'une particule ponctuelle qui tourne autour d'un axe passant par un centre
de rotation à une distance r est:
Ec =1
2mv2 (II. 1)
m: Masse de la particule. v: Vitesse de la particule.
La vitesse peut être exprimée en fonction de la vitesse angulaire ω. ω =v r (II. 2) EC = 1 2mr2ω2 = 1 2Iω2 (II. 3) Avec: I = mr2 (moment d’inertie).
La rotation d’un système est caractérisée par sa vitesse angulaire ω et par son moment d’inertie I.
Dans les molécules diatomiques chaque atome est considéré comme un point matériel de masse mA, mB. Le moment d’inertie est en général défini pour les molécules polyatomique par: I = ∑ miri2
i
(II. 4)
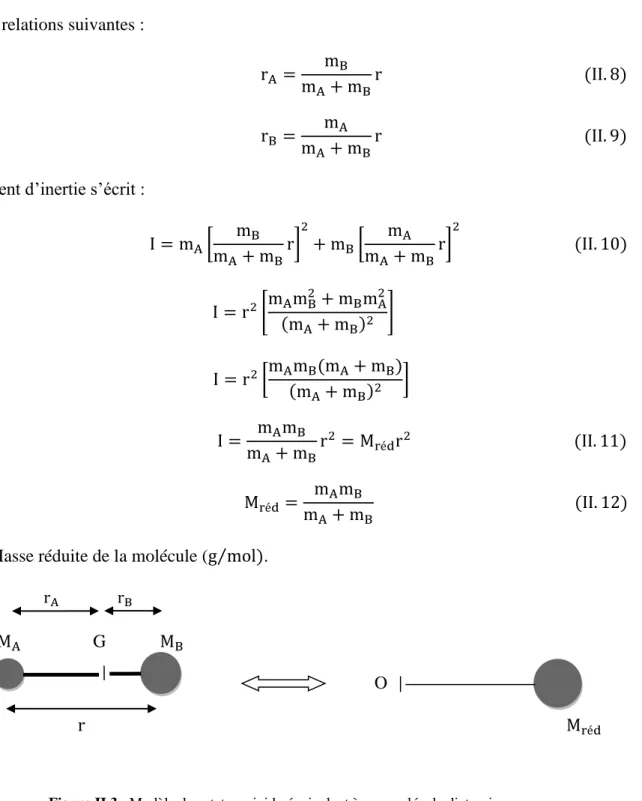
ri : La distance de la ièmeparticule de masse mi au centre de gravité G. Le moment d’inertie pour les deux atomes :
I = mArA2 + m
BrB 2 (II. 5)
rA rB
MA G MB
Chapitre II : Spectroscopie infrarouge
Page 17
Figure II.2 : Représentation schématique d’une molécule diatomique hétéronucléaire.
La position du centre de gravité d’une molécule diatomique vérifie la condition suivant :
mArA = mBrB (II. 6)
r = rA+ rB (II. 7) r : La distance intermoléculaire (longueur de liaison de la molécule diatomique) On a les relations suivantes :
rA =
mB
mA+ mBr (II. 8) rB= mA
mA+ mBr (II. 9) Le moment d’inertie s’écrit :
I = mA[ mB mA+ mBr] 2 + mB[ mA mA+ mBr] 2 (II. 10) I = r2[mAmB2 + mBmA2 (mA+ mB)2 ] I = r2[mAmB(mA+ mB) (mA+ mB)2 ] I = mAmB mA+ mBr 2 = M rédr2 (II. 11) Mréd = mAmB mA+ mB (II. 12) Mréd : Masse réduite de la molécule (g mol)⁄ .
rA rB
MA G MB
O
r Mréd
Chapitre II : Spectroscopie infrarouge
Page 18
L’énergie cinétique de système est donnée par : Ec = 1 2∑ mivi2 i (II. 13) Ec =1 2[mAvA2 + mBvB2] (II. 14) Avec : vA = ωrA (II. 15) vB= ωrB (II. 16) Ec = 1 2[mArA2 + mBrB2]ω2 Ec =1 2Mrédr2ω2 = 1 2Iω2 (II. 17) En mécanique quantique les niveaux rotationnels sont quantifiés. Après la résolution de
l’équation de Schrödinger :
HrΨr = EJΨr (II. 18) Hr= Ec = 1
2Iω2 (II. 19) Hr: Hamiltonien du système en rotation.
Ψr: Fonction d’onde rotationnelle. EJ: Energie rotationnelle (en joule).
On obtient les valeurs propres de l’énergie rotationnelle EJ :
EJ=
h2J(J + 1)
8π2I (II. 20) J: Nombre quantique de rotation qui peut prendre les valeurs 0, 1, 2 …
Si l’on considère les énergies exprimées en cm−1, il est habituel d’écrire : E̅̅̅ =J EJ hc= h 8π2IcJ(J + 1) = B̅J(J + 1) (II. 21) B̅ = h 8π2Ic (II. 22)
Chapitre II : Spectroscopie infrarouge
Page 19
E̅̅̅: Energie de rotation (en cmJ −1). B̅ : Constante de rotation (en cm−1). Pour :
J = 0 on a : E̅̅̅̅ = 0, la molécule n’est pas animée d’un mouvement de rotation. 0 J = 1 on a : E̅̅̅ = 2B. J
On remarque que l’énergie augmente avec J.
II.2.1.1Règles de sélection :
Pour qu’il y ait une transition en infrarouge entre deux niveaux rotationnel il faut que :
RJJ′ = ∫ ΨJ ∗T ΨJ′dx ≠ 0 (II. 23)
RJJ′ : Moment de transition.
En infrarouge : T = µ
µ : Moment dipolaire permanent.
On déduit :
Seules les transitions ΔJ = ±1 sont permises :
Entre un niveau J et un niveau J+1 en absorption. Entre un niveau J et un niveau J-1 en émission.
Le nombre d’onde 𝜈̅𝐽 →𝐽+1 d’une transition rotationnelle est tel que [15] :
𝜈̅J →J+1 = E̅̅̅̅̅̅ − EJ+1 ̅̅̅ (II. 24) J 𝜈̅J →J+1 = B̅ (J + 1)(J + 2) − B̅J(J + 1) 𝜈̅J →J+1 = 2B̅(J + 1) (II.25) L’écart entre deux raies successives :
∆𝜈̅ = 𝜈̅J+1 →J+2−𝜈̅J →J+1 = 2B̅ (II. 26) ∆𝜈̅ = 2B̅ (II. 27) Donc les raies dans le spectre sont équidistantes et séparées par 2B̅ . (Voir la figure II.4)
Chapitre II : Spectroscopie infrarouge Page 20 𝐄̅̅̅̅ = 𝐁̅𝐉(𝐉 + 𝟏) 𝐉 𝐉 42B 6 30B 5 20B 4 12B̅ 3 6B̅ 2 2B̅ 1 0 0 ∆J = J′− J = 1 J → J′ 0 → 1 5 → 6 2B̅ 0 2B̅ 4B̅ 6B̅ 8B̅ 10B̅ 12B̅ 𝝂̅(cm−1)
Figure II.4 : Représentation des niveaux d’énergie d’un rotateur rigide.
II.2.2 Rotateur non rigide :
En principe dans le modèle rotateur rigide l’écart entre les raies est équidistant et il n’y a pas de limite de l’énergie rotationnelle.
Mais en réalité, si la molécule est en rotation suffisamment rapide la force centrifuge sera supérieure à la force de liaison et la molécule sera dissociée. Les raies du spectre ne sont pas strictement équidistants, surtout pour les J augmente.
Chapitre II : Spectroscopie infrarouge Page 21 𝐉 𝐉 −1 𝝂̅(𝐜𝐦−𝟏) ∆𝝂̅(𝐜𝐦−𝟏) 3 2 62,584180 20,802322 20,751760 20,688647 20,613042 20,525015 20,424649 20,312041 20,187295 20,050530 4 3 83,386502 5 4 104,138262 6 5 124,826909 7 6 145,439951 8 7 165,964966 9 8 186,389615 10 9 206,701656 11 10 226,888951 12 11 246,939481
Tbleau II.1 :Spectre rotationnel partiel de HCl [1].
Donc on utilise le modèle corrigé du rotateur non rigide qui est plus conforme à la réalité : E̅̅̅= B̅J (J + 1) –D̅JJ 2(J + 1)2 (II. 28) Avec ∶ D̅ =4B̅3 𝜈̅2 (II. 29) Et: 𝜈̅ = 1 2πc√ k µ (II. 30) D̅ : Constante de distorsion centrifuge toujours positive et plus petite que B̅ , elle dépend de la rigidité de la liaison et reliée au nombre d’onde de vibration 𝜈̅. [1; 3]
𝜈̅ : Nombre d’onde de vibration.
Le nombre d’onde 𝜈̅𝐽 →𝐽+1 d’une transition rotationnelle devient : 𝜈̅J →J+1 = E̅̅̅̅̅̅ − EJ+1 ̅̅̅ J
𝜈̅J →J+1= B̅(J+1) (J + 2) –D̅(J + 1)2(J + 2)2 − B̅J (J + 1) + D̅J2(J + 1)2
Chapitre II : Spectroscopie infrarouge
Page 22
L’écart entre deux raies successives :
∆ 𝜈̅ = 𝜈̅J+1 →J+2−𝜈̅J →J+1
∆ 𝜈̅ = 2B̅ − 4D̅[(J + 1)3− (J + 2)3] (II. 32) Donc les raies dans le spectre ne sont pas équidistantes (l’écart ∆𝜈̅ diminue lorsque J augmente).
II.3 Vibration des molécules
:La spectroscopie Infrarouge permet l'étude des vibrations moléculaires. Seules les vibrations qui font varier le moment dipolaire de la molécule sont actives en infrarouges.
II.3.1 Molécule diatomique :
Pour étudier les vibrations d'une molécule diatomique A-B, on assimile la liaison entre les
deux atomes A et B par un ressort de raideur K, On définira par mA et mB les masses
respectives des atomes A et B. Pour faciliter l'étude de vibrations, on utilise le modèle physique classique de l'oscillateur
harmonique.
II.3.1.1 Oscillateur harmonique :
1) En mécanique classique [6] :
L’élongation x d’un oscillateur harmonique est proportionnelle à la force exercée F1(force de rappel).
F1 = −kx (II. 33)
x = r − r0 (II. 34)
k ∶ Constante de force de la liaison ( N/m).
k mA mB re mA mB r x
Chapitre II : Spectroscopie infrarouge
Page 23
M
Figure II.6 : Modèle de l’oscillateur équivalent à une molécule diatomique.
Mréd =mmAmB AmB
Energie de système totale = Energie cinétique + Energie potentielle
Lors de l’oscillation de la molécule, il y a continuellement transformation de l’énergie potentielle en énergie cinétique, on peut donc caractériser l’énergie du système par son énergie potentielle Ep celle-ci est liée à la force F1.
dEP
dx = −F1 = kx (II. 35) EP=
1
2kx2 (II. 36)
C’est une équation d’une parabole avec 𝑬𝒑= 𝟎 pour r = 𝐫𝟎 Pour déterminer les mouvements des atomes, On applique la seconde loi de Newton :
∑ F⃗ =Mréda⃗ (II. 37)
F⃗ : Forces extérieures agissent sur la molécule. A⃗⃗ : Accélération. Mréd d 2x dt2 = F1 (II. 38) Mréd d 2x dt2 = −kx (II. 39) La solution de cette équation différentielle de second ordre est :
x = x0cos(2π𝜈t + φ) (II. 40) 𝜈 : Fréquence de vibration(s−1). . φ: Facteur de phase. Donc: d2x dt2 = −(2π𝜈)2x (II. 41) (2π𝜈)2xM réd = kx (II. 42)
Chapitre II : Spectroscopie infrarouge
Page 24
On obtient la fréquence de l’oscillateur harmonique « loi de Hooke » :
𝜈 = 1 2π√
k
Mréd (II. 43) On peut convertir la fréquence ʋ en nombre d’onde 𝜈̅
:
𝜈̅ = 1 2πc√ k Mréd (II. 44) 𝜈̅ ∶ Le nombre d’onde (cm−1). c : La vitesse de la lumière(m s⁄
On remarque que 𝜈̅ augmente si la constante de force de liaison 𝐤 augmente et elle diminue si la masse réduite Mred augmente. La constante de force K augmente quand le nombre de liaison augmente.
La force de liaison augmente
C − C C = C C ≡ C 1200 1650 2150 cm−1 La fréquence augmente
La masse atomique augmente
C − H C − C C − O 3000 1200 1100 cm−1 La fréquence diminue
Figure II.7 : Influence de la force de liaison et de la masse atomique sur la fréquence [22].
Chapitre II : Spectroscopie infrarouge
Page 25
2) En mécanique
quantique [16] :
Selon la mécanique quantique, le mouvement de la molécule est décrit par l'équation de
Schrödinger. Dans le cas du système à une dimension x :
HvΨv = Ev Ψv (II. 45)
Hv: Hamiltonien de système. Ψv: Fonction d’onde vibrationnelle. Ev: Energie vibrationnelle.
Hv = Ec+ Ep (II. 46)
Sachant que Ec = Px 2
2Mréd (II. 47) Avec : Px = −iħ∇ (opérateur impulsion)
Et Ep= 1 2kx2 (II. 48) Avec: ω = 2πʋ ω = √ k Mréd ⟹ 𝜈 = 1 2𝜋√ k Mréd ω: Pulsation de l’oscillation sinusoïdale.
Ep = 1 2Mrédω2x2 (II. 49) L’hamiltonien H s’écrit : Hv = Px 2 2Mréd+ 1 2Mrédω2x2 (II. 50) On définit les opérateurs suivants :
Q = √Mréd ω ħ x (II. 51) P = 1 √Mrédħω px (II. 52) H1 = 1 2[Q2+ P2] (II. 53)
Chapitre II : Spectroscopie infrarouge
Page 26
H1 = Hv ħω Les opérateurs Q et P ne commutent pas.
[Q, P] = QP − PQ = i (II. 54) Avec ces notations, l’équation de Schrödinger s’écrit :
H1Ψv = Ev
ħωΨv (II. 55) On définit deux nouveaux opérateurs qui facilitent la recherche des valeurs propres de H1 : a = 1
√2(Q + iP) (II. 56) a+ = 1
√2(Q − iP) (II. 57) a : Opérateur d’annihilation. L’application de l’opérateur a fait passer le système d’un état d’énergie En à un état En−1 et ≪
détruit ≫ une quantité d’énergie ħω. En
a
En−1
a+: Opérateur de création. L’application de l’opérateur a+fait passer le système d’un état d’énergie 𝐸
𝑛 à un état 𝐸𝑛+1 , on peut dire que l’opérateur 𝒂+ ≪ crée ≫ une quantité d’énergie ħω.
En+1 a+
En On effectue le calcul du produit aa+, on obtient :
a+a = [Q − iP √2 ] [ Q + iP √2 ] (II. 58) a+a =1 2(Q2+ P2 + i(QP − PQ)) a+a = 1 2(Q2+ P2+ i[Q, P])
Chapitre II : Spectroscopie infrarouge
Page 27
a+a =1
2(Q2+ P2− 1) (II. 59) On pose : v = a+a
v : Nombre quantique de vibration qui peut prendre des valeurs entier 0,1, 2, … D’après l’équation (II.53) :
H1 = v +1
2 (II. 60) D’autre part,
Hv = ħωH1 (II. 61) A prés résolution de l’équation de Schrödinger (II.45), On déduit les valeurs propres de l’oscillateur harmonique sont données par :
Ev = ħω (v + 1 2) (II. 62) Ev = h𝜈 (v + 1 2) (II. 63) L'énergie de la molécule ne peut posséder que certaines valeurs bien définies, on parle alors des valeurs discrètes, énergie quantifiée. Pour le niveau fondamental v = 0 :
E0 = h𝜈 2
On remarque que dans l’état le plus bas d’énergie, la molécule est animée d’une certaine vibration. Les niveaux d’énergie sont équidistants. (Voir la figure II.8)
Ep 7h𝜈 2 v = 3 5h𝜈 2 v = 2 3h𝜈 2 v = 1 h𝜈 2 v = 0
x
Chapitre II : Spectroscopie infrarouge
Page 28
II.3.1.2. Les règles de sélection [1; 15] :
Pour une molécule active en IR il faut que le moment dipolaire permanent µ varie au cours
d’une transition. Une transition vibrationnelle est observée si :
le moment de transition Rvv′≠ 0
Rvv′ = ∫ Ψv∗µΨv′dx (II. 64)
µ : moment dipolaire permanent. ΨvΨv′ : Fonctions d’onde vibrationnelles à l’état inférieur et supérieur
respectivement.
Dans les molécules diatomiques homonucléaires (H2 ,O2) :Le moment dipolaire ne varie pas ∆µ = 0, car les deux atomes possèdent la même électronégativité (électronégativité c’est le pouvoir d’attirer les électrons.), ce qui
conduit à Rvv′= 0 et toutes les transitions vibrationnelles sont interdites. Dans les molécules diatomiques hétéronucléaires qui possèdent deux atomes
caractérisées par des électronégativités différentes, la densité électronique associée à la liaison n’est pas uniformément répartie, donc la molécule est polaire. Lors de la vibration de la molécule, la liaison est étirée puis comprimée donc modification du moment dipolaire et la molécule est active dans l’IR. [1]
µ
− +
r
Figure II.9 : Représentation schématique d’une molécule diatomique hétéronucléaire équivalente à un dipôle
électrique simple.
Moment dipolaire varie linéairement avec la distance internucléaire cette variation développée en série de Taylor au premier ordre:
µ = µ0+ (dµdx)
x=0x = µ0+ µ1x (II. 65)
µ
0 : Moment dipolaire en r = re .x = r − re re: Distance intermoléculaire à l’équilibre.
Il est possible d’évaluer le moment de transition en écrivant :
Chapitre II : Spectroscopie infrarouge
Page 29
Le premier terme est nul suite aux relations d’orthogonalité de la fonction d’ondes vibrationnelles. v ≠ v′ ⟹ ∫ Ψ v∗Ψv′dx = 0 (II. 67) Donc : Rvv′= µ1∫ xΨv∗Ψv′dx (II. 68) On a: Ψv(x) = (απ) 1 4 ( 1 2vv!) 1 4 e−s 2 2Hv(s) (II. 69) s = √α x Hv(s): Polynôme d’Hermite. H0(s) = 1 H1(s) = 2s sHv(s) = 1 2Hv+1(s) + vHv−1(s) (II. 70) Donc : Rvv′= µ1( α π) 1 2 ( 1 2vv!) 1 2 ( 1 2v′v′!) 1 2 ∫ e−s2xH v(s) Hv′(s)dx (II. 71)
Par changement de variable :
s = √α x ⟹ x = s √α dx = ds √α Rvv′= µ1 α ( α π) 1 2 ( 1 2vv!) 1 2 ( 1 2v′v′!) 1 2 ∫ e−s2 [1 2Hv+1(s) + vHv−1(s)] Hv′(s) ds (II. 72)
L’intégrale se compose de la somme de deux termes : 1 2∫ e−s 2 Hv+1(s)Hv′(s) ds (II. 73) v ∫ e−s2 Hv−1(s)Hv′(s) ds (II. 74)
Chapitre II : Spectroscopie infrarouge
Page 30
Le premier terme est nulle sauf si v’= v + 1. Le deuxième terme est nulle sauf si v’= v − 1. Le moment de transition n’est différent de zéro que :
∆v = v′− v = ±1 Donc, les règles de sélection sont :
∆v = ±1
Les intensités des transitions sont également proportionnelles à |Rvv′|2, elles décroissent rapidement lorsque v’ augment car la population Nv du niveau vibrationnel v est reliée à N0 par le facteur de Boltzmann :
Nv
N0 = exp (− Ev kT)
II.3.1.3 Oscillateur anharmonique :
Le modèle de l’oscillateur harmonique est très simple ne permet pas d’expliquer complètement certains phénomènes. L'énergie potentielle de l’oscillateur harmonique croît quand la distance internucléaire augmente, signifie que l'énergie potentielle est infinie pour des atomes très éloignés les uns des autres, donc il faut fournir une énergie infinie pour séparer les deux atomes. Mais on sait que cette dernière énergie est finie. C'est l'énergie de dissociation de la molécule. Donc la forme de la courbe d’énergie de la molécule en fonction du déplacement n’est pas une parabole et les niveaux ne sont plus équidistants et se rapprochent lorsque v croît. Le modèle de l’oscillateur anharmonique est plus proche de la réalité des vibrations moléculaires. Ep Ep
Oscillateur harmonique Prise en compte de l’anharmonicité Figure II.10 : Différence entre la courbe de potentiel de l'oscillateur harmonique et de l’oscillateur anharmonique.
Chapitre II : Spectroscopie infrarouge
Page 31
Pour établir un modèle mécanique anharmonique il est tout d’abord nécessaire de modéliser la courbe de l’énergie potentielle en fonction des déplacements, il y a plusieurs approches qui ont été utilisées pour trouver des modèles de courbe d’énergie qui se rapprochent de la réalité, exemple le potentielle de Morse.
Potentiel de Morse est une expression purement empirique qui suit avec une bonne
approximation de la courbe d’énergie potentielle. [2]
Ep=De[1 − exp(1 − αx)]2 (II. 75) α ∶ Constante caractéristique de la liaison entre les atomes. De: Énergie de dissociation.
L’utilisation de ce potentiel permet de construire la fonction d’Hamiltonienne. II.3.1.3.1 Oscillateur anharmonique quantique:
Le Potentiel de Morse permet de trouver des solutions presque exactes de l’équation de Schrödinger. HvΨv = EvΨv (II. 76) Hv =1 2 Px2 Mréd+ De[1 − exp(1 − αx)]2 (II. 77) On obtient les valeurs propres de l’énergie rotationnelle Ev :
Ev = h𝜈 (v +1 2) − h𝜈xe(v + 1 2) 2 + h𝜈ye(v +1 2) 3 + h𝜈ze(v +1 2) 4 + ⋯ (II. 78) Avec :
x
e, yeet ze: Constantes d’anharmonicité.On peut négliger les constantes d′anharmonicité (y
e , ze… ) pour l’étude des vibrations fondamentales parce que les valeurs des ces constantes sont [1] :
𝜈xe ≪ 𝜈 et 𝜈ye≪ 𝜈xe …
Chapitre II : Spectroscopie infrarouge Page 32 Molécules Nombre d’onde 𝝂̅ (𝐜𝐦−𝟏) Anharmonicité 𝝂̅𝐱𝐞(𝐜𝐦−𝟏) Anharmonicité 𝝂̅𝐲𝐞(𝐜𝐦−𝟏) HCl 2990 62 0,22 HBr 2649 53 -0,003 HI 2309 45 0,02
Tableau II.2 : Les constantes 𝜈̅, 𝜈̅xeet 𝜈̅ye de quelques molécules diatomiques [6].
Les valeurs d’énergie de l’oscillateur anharmonique sont données par : E̅v = 𝜈̅ (v + 12) – 𝜈̅xe(v + 12)
2
(II. 79) E̅v ∶ Energie de vibration en cm−1.
𝒙
𝒆est une valeur toujours positive, c’est pour cette raison les niveaux d’énergies ne sont pas équidistants (l’espace entre les niveaux d’énergie diminue quand 𝐯 augment et ces niveaux convergent vers une valeur limite qui corresponde à la dissociation).Ev
v
LDe v = 1 v = 0 re
Chapitre II : Spectroscopie infrarouge
Page 33
Il est impossible que la molécule s'arrête complètement de vibrer. Même dans l'état énergétique le plus bas, la molécule possédera toujours un peu d'énergie de vibration. Ce niveau d'énergie minimum est appelé énergie du point zéro. [1]
Nous avons décrit l’anharmonicité mécanique, il existe également une anharmonicité électrique dont le rôle est important pour les règles de sélection. L’anharmonicité électrique consiste à développer le moment dipolaire μ en fonction de la coordonnée interne de vibration x avec des termes d’ordre supérieur à 1 :
µ = µ0+ µ1x + µ2x2+ … (II.80) µ= µ0+ (dµdx ) x=0x + 1 2! ( d2µ dx2 )x=0 x2 + … (II.81)
L’anharmonicité électrique contribue à l’intensité des bandes fondamentales et des bandes
harmonique. Donc, l’anharmonicité mécanique modifie la position et l’intensité, mais anharmonicité
électrique modifie l’intensité. [6]
Les règles de sélection sont les même que pour l’oscillateur harmonique mais les transitions
Δv= ±2 , ±3 … sont possibles (premier ou seconde harmonique), elles sont moins intenses que les transitions Δv=±1.Toutes les transitions de l’état fondamental v = 0 à l’état excité sont permises. Le nombre d’onde ʋ0→vde la bande dûe à la transition de v = 0 → v
𝜈̅0,v = E̅v− E̅0 (II. 82) 𝜈̅0,v = 𝜈̅ (v + 12) + 𝜈̅xe(v + 12)
2
−12𝜈̅ −14𝜈̅xe
𝜈̅0,v = [1 − xe(v + 1)]v𝜈̅ (II. 83) Pour la bande fondamentale, Δv = +1 :
𝜈̅0,1 = E̅1 − E̅0 = 𝜈̅[1 − 2xe] (II. 84) Pour la première harmonique, Δv=+2:
𝜈̅0,2 = E̅2− E̅0 = 2𝜈̅[1 − 3xe] (II. 85) Pour la seconde harmonique, Δv=+3:
Chapitre II : Spectroscopie infrarouge
Page 34 La différence d’énergie entre les niveaux successifs v et v’(v’ = v+1) donnée par : 𝜈̅v,v′ = E̅v+1− E̅v 𝜈̅v,v′= 𝜈̅ (v + 32 ) +𝜈̅xe(v + 32) 2 − 𝜈̅(v + 12)− 𝜈̅xe(v + 12) 2 𝜈̅v,v′= 𝜈̅ − 𝜈̅x(2v + 2) 𝜈̅v,v′= [1 − 2(v + 1)xe] 𝜈̅ (II. 87) v peut passer de 1 →2 ou de 2 → 3 néanmoins, les probabilités des transitions de 2 →3 sont faibles, et les bandes correspondantes sont peu intenses. C’est bandes connues sous le nom de bandes chaudes.
La nomenclature des bandes vibrationnelles est explicitée sur la figure II.12.
3
2 Seconde harmonique
Premier harmonique
1 Bande chaudeBande fondamentale 0
Figure II.12 : Nomenclature des bandes de vibration.
II.3.2
Molécules polyatomiques :
Considérons une molécule composée de N atomes. 3N coordonnées sont nécessaires pour
définir la position de N atomes. Le mouvement global d’une molécule non linéaire est repéré par 6 coordonnées (3 pour la translation du centre de gravité, et 3 pour les rotations du système autour de celui-ci), et le mouvement global d’une molécule linéaire est repéré par 5
coordonnées (3 pour la translation du centre de gravité, et 2 pour les rotations parce que la rotation autour de l'axe de la molécule n'a pas de signification physique). [1]
Donc, en règle générale, le nombre de modes de vibration est :
3N- 6 pour une molécule polyatomique non linéaire. 3N- 5 pour une molécule polyatomique linéaire.
Exemple :
Chapitre II : Spectroscopie infrarouge
Page 35
3(3) − 6 = 3
La molécule CO2 est une molécule linéaire, elle présente 4 modes de vibrations. 3(3) − 5 = 4
On peut considérer une molécule polyatomique comme une structure formée des atomes reliés par des ressorts vérifiant la loi de Hooke. Ce modèle simplifié permet de comprendre, si un atome est déplacé de sa position, ce déplacement va induire dans toute la molécule un mouvement vibrationnel compliqué résultant de l’élongation des liaisons et de la déformation des angles entre celles-ci. Ces mouvements sont décomposés en modes normaux de vibration [1; 3].
On utilise la théorie de groupe pour déterminer les modes de vibration d’une molécule voir la page 39.
Un mode normal de vibration permet de considérer la molécule polyatomique comme un ensemble d’oscillateurs harmoniques indépendant qui vibrent avec lamême fréquence et qui se déplacent en phase mais avec des amplitudes différentes. Le nombre des modes normaux est égal au nombre de degrés de liberté vibrationnels.
Les vibrations d’une molécule polyatomique sont décrites en première approximation par le modèle oscillateur harmonique. Le traitement quantique de ce modèle montre que le terme vibrationnel associé à chaque mode de vibration normal i est donné par la formule (II.88) si les vibrations ne sont pas dégénérées (l’oscillateur à une dimension) [16] :
E̅̅̅̅ = 𝜈̅vi i (vi+ 12 ) (II. 88)
ῡi: Nombre d’onde vibrationnel. vi
: Nombre quantique de vibration, prendre des valeurs 0, 1, 2, …
Pour les vibrations caractérisées par un degré de dégénérescence di la formule s’écrit :E̅̅̅̅ = 𝜈̅vi i (vi+
di
2 ) (II. 89) Si la vibration est doublement dégénérée (l’oscillateur à deux dimensions).
E̅̅̅̅ = 𝜈̅vi i (vi+ 1) (II. 90)
Si la vibration est triplement dégénérée (l’oscillateur à trois dimensions).
E̅̅̅̅ = 𝜈̅vi i (vi+ 32 ) (II. 91) Bien que, en général, un mode de vibration implique le mouvement de tous les atomes dans la molécule, mais il existe des situations où les modes normaux sont localisés dans une partie seulement de la molécule. Si c’est vrai pour une liaison X−Y déterminée, on parle dans ce cas d’un nombre d’onde caractéristique pour la vibration d’élongation ou la vibration de déformation de X−Y. Ce nombre d’onde caractéristique porte le nom de nombre d’onde de
Chapitre II : Spectroscopie infrarouge
Page 36
groupement ou fréquence de groupe (les groupes d’une molécule : C=O, C-O, O-H, C-N, N-H, … etc, peuvent être excitées presque indépendamment du reste de la molécule).
Exemple :
Le groupe O−H dans l’éthanol CH3CH2OH, le mouvement de l’atome d’hydrogène du groupe OH est approximativement celui qu’il aurait s’il était attaché à une masse infinie par une liaison dont la constante de force est caractéristique d’une liaison OH.
Dans la molécule HC≡ C − CH = CH2, les constantes de forces dans les liaisons C−C, C=C et C≡C sont tout à fait différentes. Il en résulte que les élongations des liaisons sont faiblement couplées et que chaque nombre d’onde de vibration d’élongation est caractéristique d’un groupe C−C, C=C et C≡C.
Pour de nombreuses molécules, les modes normaux sont susceptibles d’induire des vibrations de squelette (couplage fort entre les mouvements d’élongation ou déformation des atomes dans une chaine ou un cycle) qui sont spécifiques de la molécule et apparaissent à des nombres
d’onde inferieurs à 1500 cm−1. La région 1500 – 3700 cm−1 correspond à la région des groupements fonctionnels (alcool,
aldéhyde, cétone, acide...). [1]
II.3.2.1 Les modes de vibration :
L’absorption infrarouge par une molécule polyatomique se traduit par deux types de
vibration :
Vibration de valence (élongation) ;
Vibration angulaire (déformation).1
) Vibration de valence (stretching) :
La distance interatomique varie sur l’axe de liaison.Symétrique Asymétrique