HAL Id: hal-02901910
https://hal-amu.archives-ouvertes.fr/hal-02901910
Submitted on 17 Jul 2020
HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Distributed under a Creative Commons Attribution| 4.0 International License
CRISP(R)ation musculaire
Océane Ballouhey, Marc Bartoli, Nicolas Levy
To cite this version:
Océane Ballouhey, Marc Bartoli, Nicolas Levy. CRISP(R)ation musculaire. médecine/sciences, EDP Sciences, 2020, 36 (4), pp.358-366. �10.1051/medsci/2020081�. �hal-02901910�
SYNTHÈSE
REVUES
médecine/sciences 2020 ; 36 : 000-0
médecine/sciences
CRISP(R)ation
musculaire
Océane Ballouhey1, Marc Bartoli1, Nicolas Levy1-3
>
Les dystrophies musculaires sont un ensemble
de pathologies musculaires rares, caractérisées
par une faiblesse et une dégénérescence
pro-gressive du muscle. Ce sont des maladies
d’ori-gine génétiques causées par la mutation d’un ou
de plusieurs gènes impliqués dans les fonctions
musculaires. Malgré des progrès significatifs
réa-lisés dans le champ des biothérapies au cours
des dernières années, il n’existe pas, à ce jour,
de traitement curatif disponible pour ces
patho-logies. Les études menées depuis la découverte
de l’outil d’édition génomique CRISPR-Cas9 ont
néanmoins permis des avancées significatives et
prometteuses dans le traitement des dystrophies
musculaires. Le système CRISPR-Cas9 permet
une édition stable et permanente du génome et
doit permettre d’éviter les traitements longs et
répétitifs. Dans cette revue, nous aborderons les
dernières avancées thérapeutiques utilisant le
système CRISPR-Cas9 dans le cadre des
dystro-phies musculaires d’origine génétique.
<
ou encore à l’âge adulte. Ce sont des maladies d’origine génétique causées par la mutation d’un ou de plusieurs gènes impliqués dans les fonctions musculaires, tels que DMD (codant la dystrophine) impli-qué dans la dystrophie musculaire de Duchenne (DMD), la dystrophie musculaire la plus connue et la plus fréquente, CAPN3 (codant la calpaïne 3) impliqué dans les dystrophies musculaires des ceintures LGMDR1 et D4 (limb girdle muscular dystrophy type R1 and D4), ou encore DYSF (codant la dysferline) impliqué dans les dysferlinopathies (LGMDR2 et myopathie de Miyoshi).
Il n’existe pas de traitement curatif pour les dystrophies musculaires. Cependant, l’identification des gènes responsables de chaque type de dystrophie musculaire et la compréhension des mécanismes qui les sous-tendent, ont permis des avancées considérables sur le plan du développement d’approches thérapeutiques. Parmi ces dernières, les principales voies explorées sont le saut d’exon utilisant des oligo-nucléotides antisens (AON), la trans-lecture de codons stop fondée sur des molécules pharmacologiques (gentamycine, TRANSLARNA ou ataluren) ou encore la thérapie génique par transfert de gène corrigé
[3]. L’enjeu de ces stratégies thérapeutiques est d’atteindre les cel-lules satellites musculaires, celcel-lules souches adultes du muscle strié squelettique logeant le long des myofibres, puisque les principales cellules qui composent le muscle sont des cellules post-mitotiques. La réparation/régénération du muscle est donc assurée par les cellules satellites. Dans le but de mettre en place une thérapie à long terme, il est nécessaire de cibler ces cellules, surtout lorsque le traitement est initié très tôt chez les enfants en pleine croissance.
Ces dernières années, un outil d’édition génomique a permis des avan-cées majeures dans les approches thérapeutiques pour les dystrophies musculaires, et semble extrêmement prometteur pour devenir un outil de premier plan dans l’arsenal thérapeutique pour les maladies géniques. Ce système CRISPR-Cas9 permet une édition stable et per-manente du génome, et doit permettre d’éviter les traitements longs et répétitifs. Les stratégies d’utilisation de cet outil d’édition
géno-1Aix Marseille Univ, Inserm, MMG, U1251, 13000 Marseille, France. 2Département de Génétique Médicale et de Biologie Cellulaire, AP-HM, Hôpital d’Enfants de la Timone, Marseille, 13000 France. 3GIPTIS, Genetics Institute for Patients Therapies Innovation and Science, 13000 Marseille, France.
Le muscle squelettique représente environ 40 % de la masse corporelle d’un individu [1]. L’intégrité de ce tissu dynamique et plastique dépend de l’équilibre établi entre la synthèse et la dégradation des protéines qui le composent. Des facteurs, tels que la nutrition, l’équilibre hormonal, l’activité physique, le stress, mais aussi de nombreuses mutations géniques fragilisent l’homéostasie du muscle.
Il existe plus de 800 pathologies monogéniques asso-ciées à une dégénérescence et une dysfonction du muscle. Parmi celles-ci, les dystrophies musculaires sont un ensemble de pathologies musculaires rares caractérisées par une faiblesse et une dégénérescence progressive du muscle : les fibres musculaires dégé-nèrent, les muscles s’atrophient progressivement et une faiblesse musculaire s’installe [2]. Ces pathologies apparaissent à tout âge : rarement dès la naissance, mais le plus souvent durant l’enfance ou l’adolescence,
2 m/s n° 4, vol. 36, avril 2020
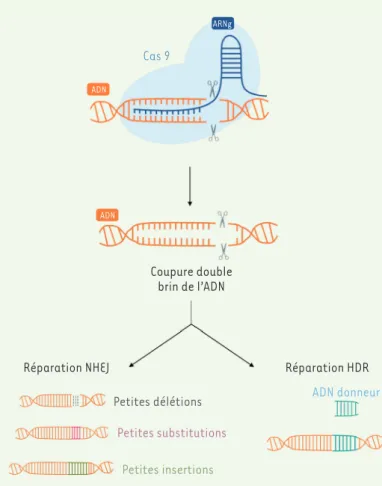
coupure double brin créée par l’enzyme Cas9 est alors réparée soit par jonction d’extrémités non-homologues (NHEJ pour non homologous end joining), résultant en de petites insertions et/ou délétions aléatoires (indel) sur le site de clivage, soit par réparation fondée sur l’homo-logie (HDR pour homology directed repair), réparation dirigée qui entraîne une modification précise du génome (Figure 1) [6]. Cependant, la précision et l’efficacité de ce système sont modulées par un inconvénient qu’il conviendra de surmonter dans l’avenir. En effet, des éditions génomiques sont susceptibles d’apparaître dans des sites adjacents d’une séquence PAM et qui ne diffèrent que de quelques nucléotides par rapport à la séquence ciblée : ces modifications sont appelées muta-tions hors cible (off-target) [7,8]. Le système CRISPR-Cas9 est ainsi capable de cliver une séquence d’ADN contenant jusqu’à 5 nucléotides qui diffèrent de l’ARN guide, ce qui est la cause de mutations hors cible. Plus le nombre de mésappariements tolérés par l’outil d’édition génomique est important, plus ce dernier générera des mutations hors cible (Figure 2).
Ces dernières années, le système CRISPR-Cas9 a large-ment été utilisé dans diverses études. Les possibilités d’utilisation de cet outil sont diverses. L’utilisation de l’enzyme Cas9 et de deux ARN guides peut permettre de déléter un fragment d’ADN, voire même un gène entier
[9]. D’un autre côté, lorsque la réparation du clivage induit par Cas9 est réalisée par le mécanisme NHEJ, il est possible d’altérer le cadre de lecture d’une protéine ou un site d’épissage. Enfin, lorsque la réparation du clivage induit par Cas9 utilise le mécanisme HDR grâce à la présence d’un ADN donneur, il est possible d’insérer un fragment d’ADN, de modifier une séquence afin de créer une mutation, ou encore de corriger une mutation. Ces utilisations diverses peuvent permettre la création de modèles dans différents organismes afin de modé-liser une pathologie. Elles permettent également de neutraliser une mutation existante afin de restaurer la production ou la fonction d’une protéine [10,11]. Pour délivrer cet outil d’édition génomique dans les organes cibles, de nombreuses méthodes ont été uti-lisées. La méthode de transport la plus utilisée in vitro est la transfection d’ADN plasmidique. In vivo, certaines études utilisent des méthodes de transport non-viral pour acheminer le système CRISPR-Cas9, telles que le transport hydrodynamique ou encore le transport par nanoparticules [12-13]. Ces stratégies de transport non-viral permettent une expression transitoire de la protéine Cas9 et ainsi de réduire les mutations hors cible. La méthode la plus utilisée pour transporter le système CRISPR-Cas9 in vivo reste le transport par AAV (virus associés aux adénovirus) [14]. Ce système viral mique sont variées. Dans cette revue, nous aborderons les dernières
avancées thérapeutiques utilisant le système CRISPR-Cas9 dans le cadre des dystrophies musculaires d’origine génétique.
Le système CRISPR
Des outils d’édition génomique existent depuis les années 1970. Ils permettent de modifier précisément une séquence génomique. Les principaux sont les nucléases à doigts de zinc (zinc finger nucleases, ZFN), les méganucléases, les nucléases effectrices de type activateur de transcription (transcription activator-like effector nucleases, TALEN) ou encore, identifié plus récemment, le système CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats) [4]. Ce dernier a per-mis des avancées considérables grâce à sa rapidité d’exécution, à sa facilité d’utilisation, et à son efficacité démontrée dans des modèles cellulaires et vivants utilisés en laboratoire. CRISPR-Cas9 est composé d’une enzyme, Cas9, et d’un ARN guide synthétique (ARNg) qui forment ensemble un complexe ribonucléoprotéique [5]. L’enzyme Cas9 chargée avec l’ARN guide reconnaît une séquence complémentaire située à côté d’une séquence spécifique de 3 à 5 nucléotides (séquence PAM, protos-pacer adjacent motif). Lorsque le complexe Cas9-ARNg reconnaît un site cible, il déclenche la dénaturation locale de l’ADN et l’invasion dans l’ADN du brin d’ARN pour assurer le clivage de la double hélice d’ADN. La A ARNg Cas 9 ADN ADN Coupure double brin de l’ADN
Réparation NHEJ Réparation HDR
Petites délétions Petites substitutions
Petites insertions
ADN donneur
SYNTHÈSE
REVUES
pathologie, avec notamment une diminution de la force musculaire et l’émergence de phénotypes histologiques dégénératifs/régénératifs dans le muscle squelet-tique, le cœur et le diaphragme. Il représente ainsi un excellent modèle de DMD. Plus récemment, en 2018, deux équipes ont également utilisé le système CRISPR-Cas9 afin d’obtenir des organismes modèles de DMD : une souris dont l’exon 50 du gène Dmd a été délétée
[16], et un lapin possédant des mutations dans l’exon 51 du même gène [17]. Enfin, un modèle de la myopa-thie a été réalisé grâce à l’outil CRISPR-Cas9 chez le singe en ciblant l’exon 4 et 46 du gène Dmd, afin d’en faire un modèle pertinent d’évaluation des stratégies thérapeutiques [18].
D’autres modèles de dystrophies musculaires ont éga-lement été créés. Une souris modélisant la dystrophie musculaire des ceintures de type R5 (LGMDR5) a ainsi été obtenue par Demonbreun et ses collaborateurs
[19]. Cette pathologie est causée par des mutations dans le gène SGCG qui code la γ-sarcoglycane. Dans le but de reproduire la mutation principale de cette dystrophie musculaire (521ΔT), les auteurs ont utilisé le système CRISPR-Cas9 afin de déléter un nucléotide de l’exon 6 du gène Sgcg, produisant un décalage du cadre de lecture et donc l’absence de production de la protéine. Une autre souris modélisant la myopathie liée au récepteur RYR1 (ryanodine receptor 1) a été réalisée [20] par Brennan et ses collaborateurs qui ont introduit une mutation ponctuelle identique à celle identifiée chez un patient, dans un allèle du gène Ryr1, ainsi qu’une délétion de 16 nucléotides produi-sant un décalage du cadre de lecture dans le deuxième allèle. Ce modèle murin présente une réduction de la masse musculaire due à une atrophie des myofibres, avec un impact sur la masse corporelle globale. L’outil CRISPR-Cas9 a donc permis, ces dernières années, de modéliser plusieurs formes de dystrophies muscu-laires, de façon plus précise, rapide et efficace que les approches utilisant la réparation par homologie permettant l’inactivation d’un gène (knock-out).
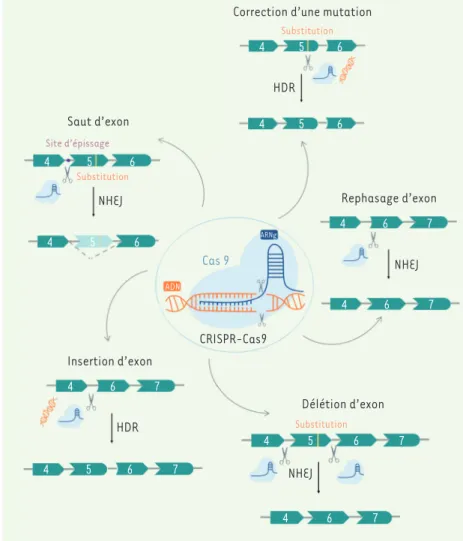
Thérapie par saut d’exon
Le système CRISPR-Cas9 ne permet pas seulement de modéliser une pathologie, il rend possible son traitement. Une des utilisations thérapeutiques du système CRISPR-Cas9 est l’altération d’un site d’épis-sage afin de réaliser un saut d’exon pour restaurer le cadre de lecture permettant de produire une protéine (Figure 3). À la différence de la thérapie par saut d’exon utilisant des oligonucléotides antisens (AON), l’utilisation de CRISPR-Cas9 permet d’altérer un site très efficace dans le muscle et versatile est fortement utilisé en
théra-pie génique, malgré sa capacité d’encapsidation assez faible.
Création d’organismes modèles
Dans le cadre des dystrophies musculaires, de nombreux modèles ont été créés en utilisant le système CRISPR-Cas9, notamment pour la myopathie de Duchenne causée par des mutations dans le gène DMD. Nakamura et son équipe ont ainsi généré un rat afin de modéliser cette pathologie [15]. Pour cela, deux exons du gène Dmd ont été ciblés dans le but de les éliminer et ainsi empêcher la production de dystro-phine. Grâce à cette modification génomique ciblée, ce rat présente de nombreuses caractéristiques phénotypiques et mécanistiques de la A ARNg Cas 9 ADN ADN ARN guide 0 mésappariement 3 mésappariements 4 mésappariements 5 mésappariements 1 mésappariement 2 mésappariements Séquence cible
Séquences non ciblées contenant 1 nucléotide différant de l’ARN guide
Séquences non ciblées contenant 2 nucléotides différant de l’ARN guide
Séquences non ciblées contenant 3 nucléotides différant de l’ARN guide
Séquences non ciblées contenant 4 nucléotides différant de l’ARN guide
Séquences non ciblées contenant 5 nucléotides différant de l’ARN guide
Figure 2. Le système CRISPR-Cas9 peut causer des mutations hors cible puisqu’il est capable de cliver une séquence d’ADN contenant jusqu’à 5 nucléotides diffé-rant de la séquence de l’ARN guide.
4 m/s n° 4, vol. 36, avril 2020
Suivant le même principe, l’équipe de Long, en 2016, a utilisé CRISPR-Cas9 chez les souris mdx, modèle standard de DMD, qui présentent une anomalie génétique dans l’exon 23 de la dystrophine, afin de réaliser le saut de l’exon 23 du gène Dmd[23]. Pour cela, ils ont transporté le système CRISPR-Cas9 grâce au virus AAV9. Leurs résultats indiquent une restauration de la dystrophine dans le muscle cardiaque et squelettique, ainsi qu’une amélioration des fonctions musculaires squelettiques (force de préhension). En 2018, Ifuku et ses collaborateurs ont altéré le site accep-teur d’épissage de l’exon 45 du gène DMD afin de provoquer le saut du même exon dans des cellules souches pluripotentes induites (iPS) dérivées d’un patient atteint de DMD dépourvu de l’exon 44 du gène DMD
[24]. Ceci a permis de restaurer le cadre de lecture et ainsi l’expression de la dys-trophine dans des myoblastes différenciés. Enfin, toujours l’équipe de Long a utilisé des ARN guides permettant d’introduire des petites insertions/délétions (indel) au niveau de 12 sites d’épissage dans des lignées d’iPS de patients atteints de DMD afin de provoquer le saut des exons correspondants, qui sont fréquemment mutés dans le gène DMD [25]. Cette étude a permis de restaurer l’expression de la dystrophine dans les cardiomyocytes dérivés d’iPS et de démontrer que la correction de 30 à 50 % des cardiomyocytes était suffisante pour restaurer le phénotype cardiaque à un niveau proche de la normale.
Thérapie par délétion d’exons
L’outil CRISPR-Cas9 a également été utilisé afin de déléter un ou plusieurs exons dans le but de restaurer la cadre de lecture d’une protéine ou de supprimer un ou plusieurs exons contenant des mutations (Figure 3). À la différence de la thérapie par saut d’exon, cette approche thérapeutique n’implique pas la machine-rie de l’épissage. Cette utilisation thérapeutique de CRISPR-Cas9 a été explorée dans le cadre des dystro-phies musculaires. Duchêne et ses collaborateurs ont ainsi restauré le cadre de lecture de la dystrophine à partir de quatre biopsies musculaires de patients atteint de DMD (chaque biopsie possédant une délétion d’un exon différent) [26]. Pour cela, ces auteurs ont utilisé l’enzyme Cas9 et deux ARN guides ciblant chacun d’épissage afin d’empêcher l’incorporation d’un exon dans l’ARN
messager (ARNm) mature par le spliceosome. Cette approche a été largement utilisée ces dernières années, particulièrement pour en évaluer la pertinence et l’efficacité dans la DMD. Dans ce contexte, les chercheurs ont ciblé une région correspondant à un site fréquent de mutations du gène DMD (exon 45 à 55). Les mutations dans cette région provoquent un décalage du cadre de lecture, constituant ainsi un fort rationnel pour l’exploration du saut d’exon dans cette dystrophie musculaire.
Ainsi, Amoasii et son équipe ont utilisé l’outil CRISPR-Cas9 dans le but de réaliser un saut de l’exon 51 du gène Dmd dans des modèles mammifères présentant une délétion de l’exon 50 : la souris ΔEx50
[16], la souris ΔEx50-Dmd-Luc [21] et le chien ΔEx50-MD [22]. La délétion de l’exon 50 du gène Dmd provoque une altération du cadre de lecture, menant alors à l’absence de production de la dystrophine. Le saut de l’exon 51 permet ainsi la restauration du cadre de lecture et la production d’une protéine plus courte mais fonctionnelle. Ces 3 études thérapeutiques ont conduit à des résultats encourageants : restauration de la dystrophine dans le muscle squelettique, le muscle cardiaque et la diaphragme.
Correction d’une mutation Substitution Substitution Saut d’exon Site d’épissage NHEJ NHEJ Rephasage d’exon ARNg Cas 9 ADN CRISPR-Cas9 Insertion d’exon HDR HDR Délétion d’exon Substitution NHEJ 4 5 6 4 5 6 4 5 6 4 5 6 4 6 7 4 6 7 4 6 7 4 5 6 7 4 5 6 4 6 7 7
SYNTHÈSE
REVUES
ont également été utilisés par Wang et son équipe, dans le but d’éliminer les répétitions toxiques du triplet CTG dans la région 3’ du gène DMPK dans des lignées iPS de patients DM1. Pour cela, des signaux de polyadé-nylation ont été insérés en amont des répétitions CTG afin d’achever prématurément la transcription. Cette stratégie thérapeutique a permis d’augmenter la préci-sion de correction par rapport à l’approche de délétion des répétitions CTG avec pour résultats, la réversion du phénotype dans les cellules souches neurales, les neu-rones du cerveau antérieur, les cardiomyocytes et les myofibres du muscle squelettique [33].
De nouveau dans le cadre de la DMD, plusieurs études montrent la correction des mutations dans le gène DMD grâce au système CRISPR-Cas9 et la présence d’un ADN donneur [34-36]. Long et al. ont ainsi utilisé cette méthode afin de corriger la mutation présente chez la souris mdx dans sa lignée germinale [34]. L’édition géno-mique de ces souris a conduit à la production d’animaux mosaïques présentant entre 2 et 100 % de correction du gène Dmd, avec une restauration partielle de la dystro-phine. Une construction Cas9 muscle-spécifique, conte-nant l’ARN guide et une région homologue à la dystrophine (ADN donneur), a également été utilisée pour corriger la mutation de la souris mdx [35]. Les muscles traités expriment alors des taux de dystrophine atteignant 70 % du taux normal, et recouvrent une force importante après l’injection intramusculaire du système CRISPR-Cas9. Son injection dans la circulation induit l’expression généra-lisée de la dystrophine dans les muscles squelettiques et cardiaques. L’utilisation d’une construction muscle-spécifique permet de limiter le risque de mutations hors cible dans les cellules non-musculaires et de minimiser le déclenchement d’une réponse immunitaire.
Matre et al. ont, eux, obtenu la restauration de la dys-trophine en ciblant l’exon 23 du gène Dmd dans des progéniteurs musculaires de souris mdx [36] avec pour résultats encourageants, l’amélioration de la proliféra-tion et de la différenciaproliféra-tion cellulaire, et la résistance des cellules au stress oxydatif et au stress du réticulum endoplasmique. De même, en utilisant CRISPR-Cas9 et un ADN donneur afin d’induire une réparation par homologie (HDR), Lee et al. ont réussi à corriger la mutation du gène Dmd chez la souris mdx [11] avec une restauration de l’expression de la dystrophine dans les tissus musculaires. Ces auteurs ont utilisé des nanopar-ticules d’or afin de transporter directement la protéine Cas9 dans les cellules, et obtenir un clivage rapide de l’ADN et réduire les mutations hors cible.
Ces stratégies thérapeutiques, qui ont en commun l’uti-lisation d’un ADN donneur, présentent des résultats pro-metteurs pour le traitement des dystrophies musculaires. l’exon 47 et l’exon 58 du gène DMD afin de déléter les exons 48 à 57
et ainsi former un exon chimérique avec la partie 5’ de l’exon 47 et la partie 3’ de l’exon 58 du gène. Cette technique a permis de restaurer l’expression de la dystrophine dans les cellules des quatre patients. L’enzyme Cas9 et deux ARN guides ciblant les introns 22 et 23 du gène Dmd ont par ailleurs été utilisés chez des souris mdx [27-29]. Cette méthode a permis d’éliminer la mutation présente dans le gène Dmd des souris mdx grâce à la délétion de l’exon 23, et l’expression de la dystrophine a pu être restaurée dans les myofibres, les cardiomyo-cytes et les cellules souches musculaires. L’expression de la protéine a persisté durant 18 mois, et les fonctions musculaires ont été partiel-lement restaurées.
Dans le cadre de la dystrophie myotonique de type 1 (DM1 ou maladie de Steinert), deux équipes ont utilisé le système CRISPR-Cas9 afin de déléter la répétition CTG, présente dans la région 3’ du gène DMPK (dystrophia myotonica 1) et à l’origine de la pathologie [30-31]. Lorsque ce triplet CTG est répété de 50 à plusieurs milliers de fois, le fonctionnement des muscles est atteint, ainsi que celui d’autres organes, selon la forme de la maladie (difficultés respiratoires, troubles cardiaques, troubles hormonaux, etc.). Dastidar et al. ont utilisé CRISPR-Cas9 afin d’éliminer ces répétitions CTG grâce à deux ARN guides dans des cellules myogéniques dérivées d’iPS de patients présentant une DM1 [30]. Leur étude montre la disparition des foyers ribonucléaires et la restauration de la localisation de MBLN1 (musc-leblind like splicing regulator 1), un régulateur d’épissage alternatif autorégulant la transcription du gène DMPK dans les cellules myogé-niques. Une étude réalisée sur des cellules musculaires dérivées d’iPS de patients atteints de DM1, et chez une souris modélisant la DM1 (la souris transgénique DMSXL1) montre qu’avec cette méthode, les foyers
de ribonucléoprotéine disparaissent et que les anomalies d’épissage dans divers transcrits sont corrigés dans les cellules musculaires déri-vées d’iPS des patients et chez la souris [31].
Thérapie par correction de mutations
Le système CRISPR-Cas9 permet aussi de corriger des mutations grâce à la présence d’un ADN donneur afin de restaurer l’intégralité d’un gène par HDR (Figure 3). Cette stratégie thérapeutique a été explorée dans plusieurs dystrophies musculaires. Selvaraj et ses collaborateurs ont, par exemple, restauré la protéine calpaïne 3, codée par le gène CAPN3, dans des myotubes dérivés de trois lignées iPS de patients atteints de dystrophie musculaire des ceintures de type R1 [32]. Pour cela, ils ont utilisé l’enzyme Cas9, un ARN guide ciblant la mutation présente dans le gène CAPN3 et un ADN donneur afin de réparer par HDR, le clivage induit par Cas9 dans ces cellules iPS mutées. Ils ont ensuite transplanté les progéniteurs myogéniques corrigés dans un modèle murin présentant un déficit immunitaire et en calpaïne 3. Cette étude a révélé le potentiel de CRISPR-Cas9 pour la thérapie génique couplée à la thérapie cellulaire. Le système CRISPR-Cas9 et un ADN donneur 1 Souris porteuses de grandes séquences génomiques humaines (45kb) contenant le gène DMPK et une
6 m/s n° 4, vol. 36, avril 2020
dont il existe plusieurs formes aux activités différentes (SaCas9, SpCas, dCas9, etc.), de l’ARN guide, ou le choix de la méthode de transport [37].
Le choix de l’enzyme Cas9 utilisée peut apporter certains avantages. La longueur de la séquence PAM diffère selon l’enzyme (3 nucléotides pour SpCas9, 5 nucléotides pour SaCas9), ce qui permet une meilleure spécificité de l’enzyme pour son substrat. Il existe également des édi-teurs de base qui convertissent un nucléotide du génome. Ce système utilise une déaminase fusionnée à l’enzyme nCas9 (Cas9 nickase) ou à l’enzyme dCas9 (Cas9 cataly-tiquement déficiente). L’avantage de cette technique est qu’elle n’induit pas de coupure de l’ADN et ne nécessite L’avantage de ces techniques est la restauration complète de la protéine,
contrairement à la thérapie par saut d’exon et par délétion d’exon que nous avons décrites. L’inconvénient de cette méthode reste la limitation de la taille de l’ADN donneur utilisable. Il n’est donc pas possible de cor-riger de larges délétions avec cette technique.
Les défis du système CRISPR
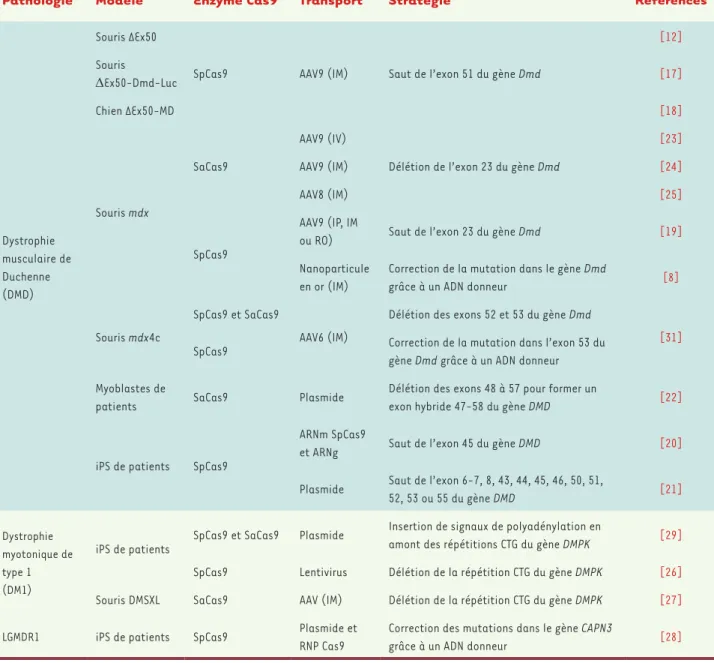
Grâce aux progrès technologiques, de nombreuses études ont été menées afin d’explorer les bénéfices thérapeutiques du système CRISPR-Cas9, notamment pour les nombreuses dystrophies muscu-laires (Tableau I). Cet outil d’édition génomique nécessite cependant de relever plusieurs défis avec en particulier, le choix de l’enzyme Cas9
Pathologie Modèle Enzyme Cas9 Transport Stratégie Références
Dystrophie musculaire de Duchenne (DMD)
Souris ΔEx50
SpCas9 AAV9 (IM) Saut de l’exon 51 du gène Dmd
[12] Souris ΔEx50-Dmd-Luc [17] Chien ΔEx50-MD [18] Souris mdx SaCas9 AAV9 (IV)
Délétion de l’exon 23 du gène Dmd
[23]
AAV9 (IM) [24]
AAV8 (IM) [25]
SpCas9
AAV9 (IP, IM
ou RO) Saut de l’exon 23 du gène Dmd [19]
Nanoparticule en or (IM)
Correction de la mutation dans le gène Dmd
grâce à un ADN donneur [8]
Souris mdx4c
SpCas9 et SaCas9
AAV6 (IM)
Délétion des exons 52 et 53 du gène Dmd
[31] SpCas9 Correction de la mutation dans l’exon 53 du
gène Dmd grâce à un ADN donneur Myoblastes de
patients SaCas9 Plasmide
Délétion des exons 48 à 57 pour former un
exon hybride 47-58 du gène DMD [22]
iPS de patients SpCas9
ARNm SpCas9
et ARNg Saut de l’exon 45 du gène DMD [20]
Plasmide Saut de l’exon 6-7, 8, 43, 44, 45, 46, 50, 51,
52, 53 ou 55 du gène DMD [21]
Dystrophie myotonique de type 1 (DM1)
iPS de patients SpCas9 et SaCas9 Plasmide
Insertion de signaux de polyadénylation en
amont des répétitions CTG du gène DMPK [29] SpCas9 Lentivirus Délétion de la répétition CTG du gène DMPK [26] Souris DMSXL SaCas9 AAV (IM) Délétion de la répétition CTG du gène DMPK [27]
LGMDR1 iPS de patients SpCas9 Plasmide et
RNP Cas9
Correction des mutations dans le gène CAPN3
grâce à un ADN donneur [28]
SYNTHÈSE
REVUES
d’anticorps spécifiques de Cas9 de bactéries parti-culières : SaCas9 (Staphylococcus aureus Cas9) et SpCas9 (Streptococcus pyogenes Cas9), les ortholo-gues les plus courants de Cas9 [44-46]. Ces résultats importants doivent donc être pris en considération à l’heure où les essais cliniques sont envisagés, afin de prévenir le risque éventuel lié à cette réponse immu-nitaire adaptative préexistante. Aucune étude ne démontre cependant que cette réponse immunitaire anti-Cas9 puisse être à l’origine d’une élimination des cellules modifiées et exprimant Cas9. L’absence d’effet de ces anticorps, qui seraient délétères pour la thérapie et limiteraient l’utilisation future chez l’homme de l’outil CRISPR-Cas9, nécessite d’être exa-minée et confirmée.
Le système CRISPR dans la société
Le système CRISPR est désormais envisagé comme outil thérapeutique pour l’homme (Tableau II) et plusieurs essais sont en cours d’évaluation. Aux États-Unis, la société Editas a ainsi obtenu l’autorisation de la FDA (Food and Drug Administration) pour effectuer un essai clinique chez des patients atteints d’Amaurose congénitale de Leber (LCA10). D’autres essais cliniques sont également réalisés pour traiter des pathologies oncologiques, telles que le myélome ou le sarcome, ou des pathologies hématologiques, telles que la drépa-nocytose et la b-thalassémie. Plusieurs défis restent néanmoins à relever avant de pouvoir utiliser cet outil comme thérapie chez l’homme. La modification du génome qu’il permet, en particulier à cause des modi-fications potentielles hors cible, peut aboutir à des effets adverses qu’il est aujourd’hui difficile d’antici-per. De telles mutations hors cible pourraient perturber l’ensemble de protéines interagissant entre elles et constituant un réseau. Or, nombre de ces interactions protéiques sont encore insuffisamment identifiées, et leur altération pourrait avoir des conséquences qui restent imprévisibles à long terme.
L’utilisation biaisée et/ou non encadrée de cet outil d’édition génomique pourrait conduire à des appli-cations en dehors du strict champ thérapeutique et à des dérives liées à sa capacité de modifier des traits non pathogènes. Sans aborder les dérives eugéniques qui pourraient résulter de cet usage, la technique CRISPR-Cas9 peut être utilisée pour modifier des carac-téristiques et, par exemple, augmenter chez l’animal, la production carnée ou en améliorer les qualités nutri-tionnelles. Il a pu être également imaginé une applica-tion humaine dans laquelle l’outil CRISPR-Cas9 pourrait être utilisé modifier certains gènes afin de doper géné-pas un ADN donneur pour la réparation de l’ADN. Dernièrement, Ryu et
son équipe ont ainsi utilisé un éditeur de base pour substituer une gua-nine à une adégua-nine, dans un modèle de souris DMD portant une mutation non-sens dans l’exon 20 du gène Dmd [38]. L’enzyme dCas9, dépourvue d’activité lytique, fusionnée avec des molécules effectrices, n’implique pas le clivage de l’ADN ciblé. Cette stratégie permet d’induire l’activation ou l’inhibition de la transcription, comme pour l’utrophine (un paralogue de la dystrophine), candidat de choix à surexprimer pour réduire le phé-notype DMD chez la souris mdx [39].
Le transport viral du système CRISPR-Cas9 par à un vecteur AAV implique plusieurs contraintes, comme la limitation de la taille d’en-capsidation du virus (4.7kb), ou la présence d’anticorps spécifiques des différents sérotypes d’AAV préexistant chez la majorité des adultes à la suite d’expositions naturelles (infection) aux AAV durant l’enfance
[13]. La taille d’encapsidation du virus est une contrainte majeure et dans la plupart des cas, les études menées sur des organismes modèles, utilisent deux transporteurs distincts : l’un pour l’enzyme Cas9, l’autre pour les ARN guides. Or, dans une optique thérapeutique, il est nécessaire de transporter l’enzyme Cas9 et les ARN guides dans le même vecteur afin d’augmenter l’efficacité de transport et de ciblage. Le vecteur AAV a également l’inconvénient de s’intégrer dans le génome de l’hôte traité, au niveau des ruptures occasionnées par l’enzyme Cas9
[40]. Les sites d’intégration du génome viral sont divers : région intro-nique (44,7 %), région intergéintro-nique (33,4 %), région exointro-nique (3,5 %) et région régulatrice (9,2 %) et il est important de prendre en compte ces insertions dans les applications d’édition génomique. Cette étude suppose aussi un potentiel rôle oncogène de l’AAV
après son intégration, due à la transactivation des gènes via la région ITR (inverted terminal repeats) du virus [51] (➜).
Le choix de la méthode de transport influence également la durée d’expression du système CRISPR dans l’organisme, avec une corréla-tion avec la fréquence d’apparicorréla-tion de mutacorréla-tions hors cible. Une étude propose une stratégie afin de limiter l’induction de ces mutations hors cible avec l’utilisation de protéines anti-CRISPR (Acr), dont l’activité est modulée par des microARN, qui permettent de limiter l’édition génomique non souhaitée dans un type cellulaire ou un tissu spécifique
[41]. Utilisant CRISPR-Cas9 et l’AcrII, qui réprime le miR122 spécifique du foie, une édition génomique spécifiquement dans le foie, et pas dans les autres tissus de l’organisme, a ainsi été obtenue.
Pour les dystrophies musculaires, l’un des défis de l’utilisation de CRISPR reste son transport ciblé au niveau du muscle et l’expression de la protéine restaurée à long terme [42-43]. Les résultats thérapeutiques précliniques obtenus montrent en effet souvent une expression de la protéine à long terme qui se révèle plus importante dans le muscle cardiaque que dans le muscle squelettique, ce qui s’explique par la régé-nération possible de ce muscle. Il est donc nécessaire que le complexe CRISPR-Cas9 atteigne les cellules satellites musculaires pour éviter la répétition des injections, mais atteindre cet objectif, des études supplé-mentaires devront être entreprises.
À noter, finalement, que des études très récentes réalisée chez l’homme ont révélé la préexistence très fréquente dans le sérum, (➜) Voir la Synthèse de A. Rossi et A. Salvetti,
m/s n° 2, février 2016,
8 m/s n° 4, vol. 36, avril 2020
genes involved in muscle function. Despite significant progress made in the field of biotherapies in recent years, there is as yet no curative treatment available for these diseases. Studies conducted since the dis-covery of the CRISPR-Cas9 genomic editing tool have nevertheless led to significant and promising advances in the treatment of muscular dystrophies. CRISPR-Cas9 system allows a stable and permanent edition of the genome and should make it possible to avoid long, partially efficient and repetitive treatments. In this review, we will discuss the latest therapeutic advances using the CRISPR-Cas9 system in genetic muscular dystrophies. ‡
REMERCIEMENTS
Nous tenons à remercier l’ensemble des membres du laboratoire MMG et de GIPTIS.
LIENS D’INTÉRÊT
Les auteurs déclarent n’avoir aucun lien d’intérêt concernant les don-nées publiées dans cet article.
RÉFÉRENCES
1. Frontera WR, Ochala J. Skeletal muscle: a brief review of structure and function. Calcif Tissue Int 2015 ; 96 : 183-95.
2. Carter JC, Sheehan DW, Prochoroff A, Birnkrant DJ. Muscular dystrophies. Clin
Chest Med 2018 ; 39 : 377-89.
3. Al-Zaidy S, Rodino-Klapac L, Mendell JR. Gene therapy for muscular dystrophy: moving the field forward. Pediatr Neurol 2014 ; 51 : 607-18.
4. Gaj T, Gersbach CA, Barbas CF. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 2013 ; 31 : 397-405.
5. Nishimasu H, Ran FA, Hsu PD, et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014 ; 156 : 935-49.
6. Jiang F, Doudna JA. CRISPR-Cas9 Structures and Mechanisms. Annu Rev
Biophys 2017 ; 46 : 505-29.
7. Zhang XH, Tee LY, Wang XG, et al. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Nucleic Acids 2015 ; 4 : 264.
8. Aryal NK, Wasylishen AR, Lozano G. CRISPR/Cas9 can mediate high-efficiency off-target mutations in mice in vivo. Cell Death Dis 2018 ; 9 : 1099.
9. Neldeborg S, Lin L, Stougaard M, Luo Y. Rapid and efficient gene deletion by CRISPR/Cas9. Methods Mol Biol 2019 ; 1961 : 233-47.
10. Min YL, Bassel-Duby R, Olson EN. CRISPR correction of Duchenne muscular dystrophy. Annu Rev Med 2019 ; 70 : 239-55.
tiquement des athlètes ou des militaires [49]2. Des gènes, tels que
MSTN (myostatin), EPOR (erythropoietin receptor), IGF1 (insulin-like growth factor 1) ou FST (follistatin) pourraient ainsi être ciblés : par exemple, une mutation du gène MSTN provoque une hypertrophie mus-culaire [50]. Josiah Zayner, un chercheur en biophysique moléculaire et qui a travaillé pour la NASA, a ainsi tenté en 2017 de modifier une partie de son génome dans le but d’augmenter sa masse musculaire en s’injectant l’enzyme Cas9 et un ARN guide ciblant le gène MSTN afin d’empêcher la production de myostatine, qui inhibe la croissance musculaire. Cette expérience fut un échec notoire mais elle montre que l’outils, simple d’utilisation et peu coûteux,
le rend accessible à tous scientifiques, mais aussi aux amateurs éclairés de biologie [52] (➜).
Malgré son échec, Zayner a créé une société, The ODIN, qui commercia-lise des kits permettant de réacommercia-liser de l’édition génomique à domicile, notamment en utilisant l’outil CRISPR. Cette situation a conduit les autorités fédérales et la FDA à rappeler que l’utilisation de CRISPR chez l’homme devait faire l’objet d’une autorisation, et que la vente de ces kits était illégale…
Face aux risques liés à de telles situations et à la rapidité de déve-loppement de cette technologie, en termes d’efficacité, de précision, de ciblage, de simplification et de reproductibilité, il semble donc essentiel et urgent de définir les limites de son utilisation, ses indica-tions et qu’un cadre soit établi par les autorités sanitaires. C’est une des conditions indispensables pour que la technologie accompagne la médecine et la science et grâce à laquelle les meilleurs bénéfices thérapeutiques seront apportés aux malades, sans risques de dérive sociétale. ‡
SUMMARY
CRISPR-Cas9 for muscle dystrophies
Muscular dystrophies are a group of rare muscular disorders charac-terized by weakness and progressive degeneration of the muscle. They are diseases of genetic origin caused by the mutation of one or more
2 Le dopage génétique est défini comme un transfert d’ADN ou l’utilisation de cellules génétiquement
modifiées dans le but d’améliorer une performance physique.
Société / Sponsor Désigna-tion Pathologies Méthode Phase de l’essai clinique
Editas EDIT-101 Amaurose congéni-tale de Leber (LCA10)
Édition génomique in vivo par délétion de la mutation IVS26 présente dans le gène CEP290
Phase I/II
CRISPR Therapeutics et
Vertex Pharmaceuticals CTX001
Drépanocytose Édition génomique ex-vivo des cellules souches hématopoïétiques par correction des mutations dans le gène des b-globines
Phase I/II b-thalassémie
Université de Pennsylvanie HLA-A*0201
Myélome Édition génomique ex-vivo des lymphocytes T par inhibition de l’expression du récepteur TCR et de PD-1
Phase I Sarcome
Tableau II. Études cliniques utilisant le système CRISPR-Cas9 pour traiter des pathologies chez l’homme.
(➜) Voir le Repères de M. Meyer, m/s n° 5, mai 2018, page 473
SYNTHÈSE
REVUES
33. Wang Y, Hao L, Wang H, et al. Therapeutic genome editing for myotonic dystrophy type 1 using CRISPR/Cas9. Mol Ther 2018 ; 26 : 2617-30.
34. Long C, McAnally JR, Shelton JM, et al. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 2014 ; 345 : 1184-8.
35. Bengtsson NE, Hall JK, Odom GL, et al. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat Commun 2017 ; 8 : 14454.
36. Matre PR, Mu X, Wu J, et al. CRISPR/Cas9-based dystrophin restoration reveals a novel role for dystrophin in bioenergetics and stress resistance of muscle Pprogenitors. Stem Cells 2019 ; 37 : 1615-28.
37. Hwang J, Yokota T. Recent advancements in exon-skipping therapies using antisense oligonucleotides and genome editing for the treatment of various muscular dystrophies. Expert Rev Mol Med 2019 ; 21 : e5.
38. Ryu SM, Koo T, Kim K, et al. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat Biotechnol 2018 ; 36 : 536-9.
39. Ricotti V, Spinty S, Roper H, et al. Safety, tolerability, and pharmacokinetics of SMT C1100, a 2-Arylbenzoxazole utrophin modulator, following single- and multiple-dose administration to pediatric patients with Duchenne muscular dystrophy. PLoS One 2016 ; 11 : e0152840.
40. Hanlon KS, Kleinstiver BP, Garcia SP, et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat Commun 2019 ; 10 : 4439.
41. Lee J, Mou H, Ibraheim R, et al. Tissue-restricted genome editing in vivo specified by microRNA-repressible anti-CRISPR proteins. RNA 2019 ; 25 : 1421-31.
42. Min YL, Bassel-Duby R, Olson EN. CRISPR correction of Duchenne muscular dystrophy. Annu Rev Med 2019 ; 70 : 239-55.
43. Cohen J. In dogs, CRISPR fixes a muscular dystrophy. Science 2018 ; 361 : 835.
44. Attenello FJ. Immunity to CRISPR-Cas9. Sci Transl Med 2019 ; 1 : 5328.
45. Crudele JM, Chamberlain JS. Cas9 immunity creates challenges for CRISPR gene editing therapies. Nat Commun 2018 ; 9 : 3497.
46. Charlesworth CT, Deshpande PS, Dever DP, et al. Identification of preexisting adaptative immunity to Cas9 proteins in humans. Nat Med 2019 ; 25 : 249-54.
49. Neuberger EWI, Simon P. Gene and cell doping: the new frontier. Beyond myth or reality. Med Sport Sci 2017 ; 62 : 91-106.
50. Schuelke M, Wagner KR, Stolz LE, et al. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med 2004 ; 350 : 2682-8.
51. Rossi A, Salvetti A. Intégration des vecteurs AAV et mutagenèse insertionnelle. Med Sci (Paris) 2016 ; 32 : 167-74.
52. Meyer M. Biologie et médecine « do-it-yourself ». Histoire, pratiques, enjeux. Med Sci (Paris) 2018 ; 34 : 473-9.
RÉFÉRENCES
11. Gee P, Xu H, Hotta A. Cellular reprogramming, genome editing, and alternative CRISPR Cas9 technologies for precise gene therapy of Duchenne muscular dystrophy. Stem Cells Int 2017 ; 8765154.
12. Chen G, Abdeen AA, Wang Y, et al. A biodegradable nanocapsule delivers a Cas9 ribonucleoprotein complex for in vivo genome editing. Nat Nanotechnol 2019 ; 14 : 974-80.
13. Lee K, Conboy M, Park HM, et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat Biomed Eng 2017 ; 1 : 889-901.
14. Lau CH, Suh Y. In vivo genome editing in animals using AAV-CRISPR system: applications to translational research of human disease. F1000Res 2017 ; 6 : 2153.
15. Nakamura K, Fujii W, Tsuboi M, et al. Generation of muscular dystrophy model rats with a CRISPR/ Cas system. Sci Rep 2014 ; 4 : 5635.
16. Amoasii L, Long C, Li H, et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci Transl Med 2017 ; 9.
17. Sui T, Lau YS, Liu D, et al. A novel rabbit model of Duchenne muscular dystrophy generated by CRISPR/Cas9. Dis Model Mech 2018 ; 11.
18. Chen Y, Zheng Y, Kang Y, et al. Functional disruption of the dystrophin gene in rhesus monkey using CRISPR/Cas9. Hum Mol Genet 2015 ; 24 : 3764-74.
19. Demonbreun AR, Wyatt EJ, Fallon KS, et al. A gene-edited mouse model of Limb-Girdle muscular dystrophy 2C for testing exon skipping. Dis Model Mech 2019 ; 13. pii: dmm040832.
20. Brennan S, Garcia-Castaneda M, Michelucci A, et al. Mouse model of severe recessive RYR1-related myopathy. Hum Mol Genet 2019 ; 28 : 3024-36.
21. Amoasii L, Li H, Zhang Y, et al. In vivo non-invasive monitoring of dystrophin correction in a new Duchenne muscular dystrophy reporter mouse. Nat Commun 2019 ; 10 : 4537.
22. Amoasii L, Hildyard JCW, Li H, et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 2018 ; 362 : 86-91.
23. Long C, Amoasii L, Mireault AA, et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016 ; 351 : 400-3.
24. Ifuku M, Iwabuchi KA, Tanaka M, et al. Restoration of dystrophin protein expression by exon skipping utilizing CRISPR-Cas9 in myoblasts derived from DMD patient iPS cells. Methods Mol Biol 2018 ; 1828 : 191-217.
25. Long C, Li H, Tiburcy M, et al. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci Adv 2018 ; 4 : eaap9004.
26. Duchêne BL, Cherif K, Iyombe-Engembe JP, et al. CRISPR-induced deletion with SaCas9 restores dystrophin expression in dystrophic models in vitro and in vivo. Mol Ther 2018 ; 26 : 2604-16.
27. Nance ME, Shi R, Hakim CH, et al. AAV9 edits muscle stem cells in normal and dystrophic adult mice. Mol Ther 2019 ; 27 : 1568-85.
28. Tabebordbar M, Zhu K, Cheng JKW, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016 ; 351 : 407-11.
29. Nelson CE, Hakim CH, Ousterout DG, et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016 ; 351 : 403-7.
30. Dastidar S, Ardui S, Singh K, et la. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res 2018 ; 46 : 8275-98.
31. Lo Scrudato M, Poulard K, Sourd C, et al. Genome editing of expanded CTG repeats within the human DMPK gene reduces nuclear RNA foci in muscle of DM1 mice. Mol Ther 2019 ; 27 : 1372-88.
32. Selvaraj S, Dhoke NR, Kiley J, et al. Gene correction of LGMD2A patient-specific iPSCs for the development of targeted autologous cell therapy. Mol Ther 2019 ; 27 : 2147-57.
TIRÉS À PART