Computational investigation of pathogen evolution
Texte intégral
Figure

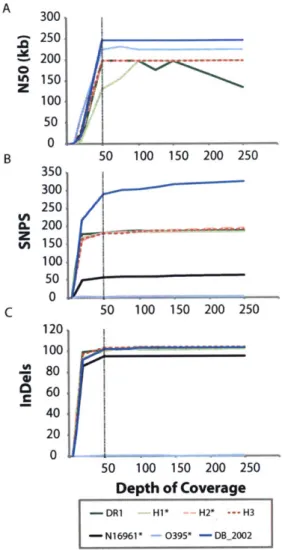
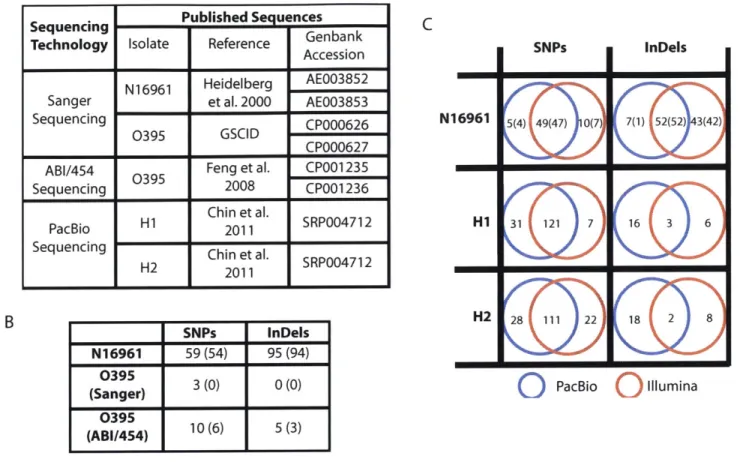


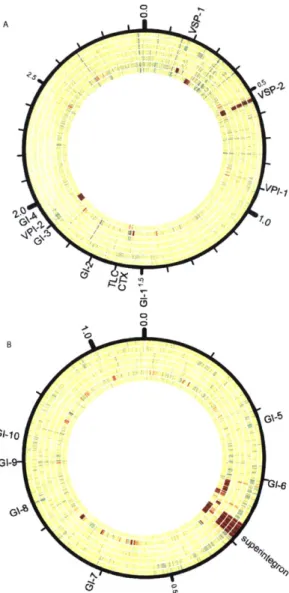

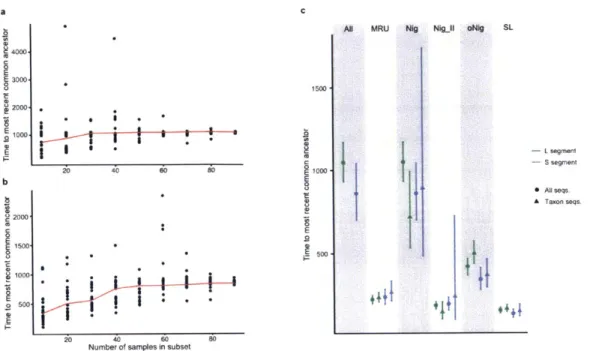

Documents relatifs
Mesoproterozoic to early Neoproterozoic metamorphosed mafic and unmetamorphosed potassic to ultrapotassic rocks from the Canyon domain, part of the polycyclic
C’était le moment choisi par l’aïeul, […] pour réaliser le vœu si longtemps caressé d’ accroître son troupeau que les sècheresses, les épizoodies et la rouerie de
Finally, as stated by the WHO position paper on cholera vaccines 1 and suggested by our additional analysis of Sévère and others data, WASH activities remain the corner stone of
Our results show that the risk of epidemic onset of cholera in a given area and the initial intensity of local outbreaks could have been anticipated during the early days of the
The field val- ues corresponding to the lower and the upper boundary of the plateau are in excellent agreement with the NMR data: at very low temperature, the coexistence between
In conclusion, we show in this thesis how evolutionary robotics can contribute to a same problem (in our case the evolution of cooperation) in two very different directions:
(2009) [26] investigated the statistical properties of random directional wave fields in the finite depth case using the HOS method [3].. Wave focussing on water of finite depth
Here, we develop a model examining the evolution of MDR in a structured host population that complements existing models in these aspects: we explicitly model the dynamics of
