Hydrogen Atom Transfer (HAT) Processes Promoted by the
Quinolinimide
‑N‑oxyl Radical. A Kinetic and Theoretical Study
Gino A. DiLabio,
†Paola Franchi,
§Osvaldo Lanzalunga,
*
,‡Andrea Lapi,
‡Fiorella Lucarini,
∥Marco Lucarini,
§Marco Mazzonna,
‡Viki Kumar Prasad,
†and Barbara Ticconi
‡†Department of Chemistry, University of British Columbia, Okanagan, 3247 University Way, Kelowna, British Columbia, Canada
V1V 1V7
‡Dipartimento di Chimica, Sapienza Università di Roma and Istituto CNR di Metodologie Chimiche (IMC-CNR), Sezione
Meccanismi di Reazione, c/o Dipartimento di Chimica, Sapienza Università di Roma, P. le A. Moro, 5, I-00185 Rome, Italy
§Dipartimento di Chimica“G. Ciamician”, Università di Bologna, Via San Giacomo 11, I-40126 Bologna, Italy ∥Département de Chimie, Université de Fribourg, Chemin du Musée 9, 1700 Fribourg, Switzerland
ABSTRACT: A kinetic study of the hydrogen atom transfer (HAT) reactions from a series of organic compounds to the quinolinimide-N-oxyl radical (QINO) was performed in CH3CN. The HAT rate constants are significantly higher than those observed with the phthalimide-N-oxyl radical (PINO) as a result of enthalpic and polar effects due to the presence of the N-heteroaromatic ring in QINO. The relevance of polar effects is supported by theoretical calculations conducted for the reactions of the two N-oxyl radicals with toluene, which indicate that the HAT process is characterized by a significant degree of charge transfer permitted by the π-stacking that occurs between the toluene and the N-oxyl aromatic rings in the transition state structures. An increase in the HAT reactivity of QINO was observed in the presence of 0.15 M HClO4and 0.15 M Mg(ClO4)2 due to the protonation or complexation with the Lewis acid of the pyridine nitrogen that leads to a further decrease in the electron density in the N-oxyl radical. These results fully support the use of N-hydroxyquinolinimide as a convenient substitute for N-hydroxyphthalimide in the catalytic aerobic oxidations of aliphatic hydrocarbons characterized by relatively high C−H bond dissociation energies.
■
INTRODUCTIONThe use of N-hydroxyphthalimide (NHPI) as a catalyst in the aerobic oxidative functionalization of hydrocarbons has attracted a great deal of attention in recent years. In the presence of metal and non-metal co-catalysts, the NHPI/O2 system was found to efficiently catalyze the introduction of oxygenated groups in aliphatic and alkylaromatic hydro-carbons.1
The key role played in the catalytic cycle by the hydrogen atom transfer from the organic substrate to the phthalimide-N-oxyl radical (PINO) [HAT process (Scheme 1)] has stimulated several kinetic studies aimed at obtaining quantitative information about the reactivity of PINO and other structurally
related short-lived N-oxyl radicals toward a variety of C−H bonds.2,3
The HAT reactivity of short-lived N-oxyl radicals is strongly dependent on enthalpic effects based on the difference between the O−H bond dissociation energy (BDE) in the N-hydroxy derivative and the C−H BDE of the substrates (Scheme1). In this respect, the high reactivity of PINO is associated with a relatively high NO−H BDE in NHPI (87 kcal/mol).4
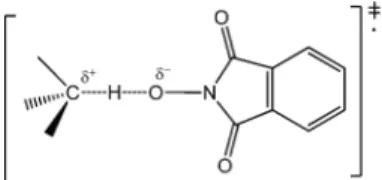
In addition to enthalpic effects, polar effects also play an important role in the HAT reactivity of N-oxyl radicals.6 The HAT transition state (TS) structure is characterized by a partial degree of charge transfer from the substrate to the N-oxyl radical (Figure 1). The polar contribution is clearly dependent on the ability of the substrate and the N-oxyl radical to stabilize the positive and negative charges, respectively, developing in the TS.
Scheme 1
http://doc.rero.ch
Published in "The Journal of Organic Chemistry 82(12): 6133–6141, 2017"
which should be cited to refer to this work.
On the basis of enthalpic considerations, an increase in the HAT reactivity of N-oxyl radicals can be established by an increase in the BDENO−H values of the corresponding N-hydroxy derivatives. In aryl-substituted N-N-hydroxyphthalimides and other aromatic hydroxylamines, an increase in BDENO−H values is observed with electron-withdrawing aryl substituents that destabilize the resonance structure with charge separation shown inFigure 2.7
Electron-withdrawing substituents also can increase the reactivity of the N-oxyl radical through polar effects on the HAT TS structure as a result of enhanced stabilization of the partial negative charge that develops on the N-oxyl radical at the TS.8 The enhancement of HAT reactivity exerted by electron-withdrawing substituents on N-oxyl radicals has been reported in several kinetic and product studies of the reaction of aryl-substituted PINO and benzotriazole-N-oxyl radicals with alkylaromatics and benzylic alcohols.7,9
The decrease in the PINO electron density can also be affected by replacement of the phenyl with an electron-withdrawing N-heteroaromatic ring [N-hydroxyquinolinimide (NHQI) (Figure 3)]. The corresponding N-oxyl radical
(QINO) should be more reactive than PINO in HAT processes, and therefore, NHQI may serve as a better catalyst than NHPI in the oxidations of organic compounds using molecular oxygen as the terminal oxidant. Via this connection, Xia et al. showed that the catalytic activity of NHQI in the aerobic oxidation of toluene is significantly higher than that of NHPI either in the absence or in the presence of a metal co-catalyst.10 More recently, it was reported that NHQI, in combination with 4-carboxyl-N-hydroxyphthalimide, showed excellent activity in the aerobic oxidation of ethylbenzene.11
In this study, we report the ultraviolet−visible (UV−vis) and electron paramagnetic resonance (EPR) spectroscopic charac-terization of QINO and a kinetic investigation of the HAT reactivity of QINO with a series of organic substrates, including
aliphatic hydrocarbons, alkylaromatics, alcohols, ethers, alde-hydes, and amides. The results were compared with those found in the HAT processes promoted by PINO. Theoretical calculations using a density functional theory (DFT)-based technique were performed for the HAT reactions of PINO and QINO with toluene, selected for the industrial relevance of the HAT from alkylaromatics to N-oxyl radicals,1 to gain insight into the energetics of the HAT process involving these species. Because an increase in the QINO reactivity of the HAT process may result from the protonation or complexation with metal cations of the heteroaromatic nitrogen, kinetic studies have been also performed in the presence of Brønsted and Lewis acids [HClO4and Mg(ClO4)2] employed at a concentration of 0.15 M.
■
RESULTSGeneration and Characterization of QINO. QINO was generated in a quartz cuvette by adding a 0.5 M solution of cerium(IV) ammonium nitrate in acetonitrile to a 1.0 M solution of NHQI at 25°C, after which the UV−vis spectrum was recorded. This method was used previously to characterize spectroscopically PINO and other transient N-oxyl radicals (Figure 4).3,12
The UV−vis spectrum so obtained has an absorption band centered at 380−390 nm. This band overlaps with the absorbance region of cerium salts (see Figure S1). Better UV−vis spectroscopic characterization of QINO was achieved via a laser-flash photolysis (LFP) experiment as described in
Scheme 2.13,14Cumyloxyl radical (CumO•), generated by LFP
at 355 nm of a 1 M solution of dicumyl peroxide in N2 -saturated acetonitrile at 25°C, abstracts a hydrogen atom from NHQI (5 mM), yielding QINO (eq 2 in Scheme 2).15This hydrogen atom abstraction reaction occurs in competition with the β-scission of CumO•, which leads to the formation of acetophenone and a methyl radical (eq 3 inScheme 2; kβ∼ 6.5 × 105 s−1 in CH
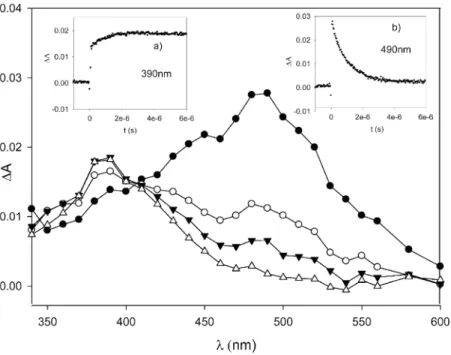
3CN).13 Figure 5 shows the time-resolved spectra obtained. Thefirst-order decay of the cumyloxyl radical recorded at its maximal absorption wavelength (485 nm)14is accompanied by the buildup of the QINO absorption at 390 nm.
Figure 1. Polar transition state in the HAT process promoted by PINO.
Figure 2.Resonance structures of aryl-substituted PINO radicals.
Figure 3. Structures of N-hydroxyphthalimide (NHPI), N-hydrox-yquinolinimide (NHQI), phthalimide-N-oxyl radical (PINO), and quinolinimide-N-oxyl radical (QINO).
Figure 4. Generation of QINO by oxidation of NHQI with cerium(IV) ammonium nitrate.
Scheme 2
The electron spin resonance spectrum of QINO was recorded at 298 K by UV photolysis of a CH3CN solution containing 10 mM NHQI and 10% (v/v) di-tert-butyl peroxide. The spectrum (seeFigure 6) shows three main lines due to the
coupling of the unpaired electron with nitroxidic nitrogen (aN= 4.57 G).16Each line also shows unresolved hyperfine structure due to weak coupling of the unpaired electron with the nitrogen and hydrogen aromatic nuclei. A similar EPR spectrum of QINO was obtained by oxidation of NHQI with cerium(IV) ammonium nitrate (CAN) in CH3CN at 25 °C (see Figure S2). The second-derivative spectrum clearly showed unresolved hyperfine structure due to weak coupling of the unpaired electron with the nitrogen and hydrogen aromatic nuclei.
NO−H Bond Dissociation Enthalpies (BDEs) of NHQI. The BDENO−H of NHQI is a parameter of fundamental importance for the analysis of the HAT reactivity of QINO. Its value, determined both experimentally and by theoretical
calculations, was compared with the BDENO−H value of NHPI previously determined with the same methodologies.2c,7a,d,8,17 The experimental determination of the O−H BDE was performed by the EPR radical equilibration technique. For this purpose, we measured the equilibrium constant (Keq) for the hydrogen atom transfer reaction between NHQI and a NHPI derivative whose O−H BDE was already known (Scheme 3). As a reference, we chose 4-CH3OCO-NHPI, which is characterized by an O−H BDE of 87.8 kcal/mol.18
The initial concentration of N-hydroxyimides was used to calculate Keq, while the relative radical concentration was determined by EPR spectroscopy (see details in theSupporting Information and Figure S3).19 On the assumption that the entropic term can be neglected, BDENHQI = BDE4‑CH3OCO‑NHPI
− RT ln(Keq). From the experimentally determined radical concentrations, we obtained a Keqof 1.20, from which an O−H BDE of 87.7± 0.6 kcal/mol for NHQI was calculated.20
Theoretical Calculations. DFT-based calculations were performed to model the reactions of QINO and PINO radicals with toluene. The B3LYP/6-31+G(2d,2p)21level of theory was used as the underlying method. The effects of noncovalent interactions in the reacting systems were modeled with the dispersion-correcting potentials (DCPs) approach described previously.22Although a number of more common approaches to correcting for the absence of dispersion in the B3LYP (and other) methods exist, we elected to use the DCP approach to obtain results comparable with those previously published for computational studies of the reactions between PINO and phenols. The method we applied is abbreviated as B3LYP-DCP/6-31+G(2d,2p).
Figure 5.Transient absorption spectra measured 98 ns (●), 288 ns (○), 1μs (▼), and 2μs (△) after 355 nm laser excitation of a solution of dicumyl peroxide (1 M) and NHQI (5 mM) in CH3CN at 25°C under N2. Inset a shows thefirst-order buildup of the QINO absorption at 390 nm.
Inset b shows thefirst-order decay of the absorption of the cumyloxyl radical at 490 nm.
Figure 6. EPR spectrum of QINO recorded at 298 K during UV photolysis of a CH3CN solution containing 10 mM NQHI and
di-tert-butyl peroxide [10% (v/v)].
Scheme 3
We computed the energy-optimized structures of the reactants, pre- and postreaction (noncovalently bound) complexes, and the cisoid (π-stacked) and transoid (non-stacked) transition state (TS) structures in an implicit solvent field (acetonitrile) using the SMD model of Marenich et al.23
All non-TS structures were verified to have only positive vibration frequencies, and the TS structures had a single imaginary frequency that led to the corresponding pre- and postreaction species. The gas- and solvent-phase energy-optimized Cartesian coordinates of the reactants, pre- and postreaction complexes, transition states, and products are provided in theSupporting Information.
The O−H BDEs for NHQI and O−H BDEs for NHPI were computed using the restricted open-shell (RO) version of CBS-QB3.24All calculations were performed using the Gaussian-09 program package.25
The (RO)CBS-QB3 method predicts the gas-phase O−H BDEs in NHQI and NHPI to be 84.0 and 83.1 kcal/mol, respectively,26with a difference of 0.9 kcal mol−1, which is in line with the experimentally determinedΔBDENO−H(0.7 kcal/ mol). The same approach predicts the C−H BDE of the methyl group in toluene to be 89.7 kcal/mol. For comparison, the B3LYP-DCP/6-31+G(2d,2p) approach predicts the analogous BDEs in NHQI, NHPI, and toluene to be 80.0, 79.0, and 88.8 kcal/mol, respectively. We attribute the lack of agreement between the (RO)CBS-QB3 and B3LYP-DCP BDEs for NHQI and NHPI to delocalization errors associated with the underlying B3LYP functional. This suggests that the B3LYP-derived barrier heights and reaction endergonicity will be higher than the actual values (see below).
The calculated solvent-phase free energies associated with the reaction of QINO and PINO with toluene in acetonitrile solvent are listed in Table 1. In the gas phase, the reactants
form fairly strongly bound pre-reaction complexes, with enthalpies of association of 8.4 kcal/mol for QINO and 7.8 kcal/mol for PINO. However, the inclusion of entropic correction results in pre-reaction complex formation processes that are endergonic [2.6 kcal/mol in both cases (seeTables S1 and S2)]. The inclusion of estimates of the solvent effects
increases the endergonicity of pre-reaction complex formation to 5.5 kcal/mol for QINO and 4.6 kcal/mol for PINO. These results are consistent with both the relatively high polarity of the radicals and the differential in the polarity between the radicals.
The TS structures associated with the HAT reaction of QINO and PINO with toluene were found to beπ-stacked, or “cisoid” (Figure 7), rather than a “transoid” structure [i.e., nonstacked, or open (see Figure S43)]. This finding is
consistent with the calculated structure for the HAT identity reaction for the toluene/benzyl27 and phenol/phenoxyl28 couples. As was discussed in ref8, the overlap of the aromatic moieties results in some degree of noncovalent bonding interaction between them within the TS structure, thereby stabilizing the cisoid TS structure preferentially with respect to the transoid structure. In the case of the reactions of QINO and PINO with toluene, the free energies of the cisoid transition state structures are 3.2 kcal/mol lower than those of the transoid structures in the gas phase (see Tables S1 and S2). The preferential stabilization of the cisoid TS structures persists in solvent, but the differential between the cisoid and transoid structures is reduced by∼1 kcal/mol (seeTables S3 and S4), presumably because the transoid TS structures are more stabilized by interactions with the solventfield.
The calculated free energy barrier height for the reaction between QINO and toluene is 19.6 kcal/mol. As expected on the basis of the arguments made above, this barrier is found to be 1 kcal/mol lower than that associated with the PINO reaction. Both barriers are expected to be overestimated by approximately 2−3 kcal/mol on the basis of the fact that B3LYP predicts a larger differential between the NHQI and NHPI O−H BDEs and the toluene methyl group C−H BDE than does (RO)CBS-QB3.
The lower free energy barrier in the QINO−toluene HAT reaction is consistent with the fact that there is a higher degree of charge transfer between the reactants in this system than in the PINO−toluene reaction. For example, our solvent-phase calculations indicate that 0.47 electron is transferred from toluene to the QINO−toluene complex in the cisoid TS complex, which is an additional 0.06 electron compared to the number in the analogous PINO−toluene complex. For comparison, the degree of charge transfer in the transoid TS structures is 2−4-fold lower. This supports the notion that the ring stacking interactions in the TS facilitate charge transfer and promote HAT.
After the hydrogen atom transfer is completed, the calculations predict that the species involved in the reaction will not remain complexed in acetonitrile. The enthalpies and free energies associated with complexation within the post-reaction complex are predicted to be +3.4 and +13.3 kcal/mol for NHQI-benzyl and +4.7 and +14.4 kcal/mol for NHPI-benzyl, respectively, relative to the reactants in solvent. The free energies of thefinal dissociated products of the HAT reaction in solvent are 10.3 kcal/mol (QINO) and 11.4 kcal/mol (PINO) higher than those of the reactants. On the basis of the differences between the B3LYP and (RO)CBS-QB3 bond dissociation energies, our expectation is that the free energies of the two reactions are closer to 7−8 kcal/mol.
Kinetic Study of the HAT from C−H Bonds to QINO and PINO. Kinetic studies were performed by UV−vis spectrophotometry generating QINO and PINO by oxidation of NHQI and NHPI with cerium(IV) ammonium nitrate in CH3CN at 25°C.3,12,29 The decay of the N-oxyl radicals was recorded at 390 and 380 nm for QINO and PINO, respectively. Using an excess of substrates (at least 10-fold) to attain pseudo-first-order conditions, clean pseudo-first-order decays were observed with the H-abstraction rate being faster than the spontaneous decay of the N-oxyl radicals and excellent linear fits were obtained by plotting the pseudo-first-order rate constants (kobs) as a function of the concentration of hydrogen donors. From the slope of these plots, the second-order rate constants for Table 1. Reaction Free Energies, Relative to Reactants, of
the Hydrogen Atom Transfer from Toluene to QINO and PINO in an Acetonitrile Solventa
reaction step QINO with toluene PINO with toluene pre-reaction complex 5.5 4.6 cisoid TS complex 19.6 20.6 transoid TS complex 21.8 23 postreaction complex 13.3 14.4 products 10.3 11.4
aThe data were obtained using the B3LYP-DCP/6-31+G(2d,2p)
approach with the SMD solvation (acetonitrile) model. All values are in kilocalories per mole.
HAT (kH) were determined, as shown in Figure 8 for the reactions of QINO with alkylaromatics.30
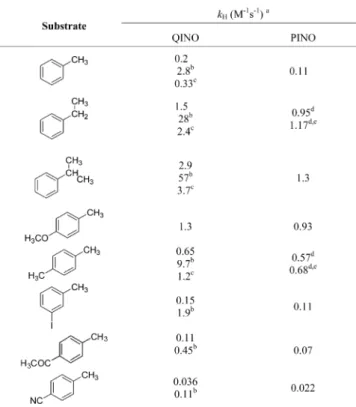
The kHvalues are listed inTable 2for the reactions of QINO and PINO with alkylaromatics and in Table 3 for the other hydrogen atom donors. In Table 2 are also reported the kH values for the HAT reactions of QINO with alkylaromatics in the presence of either 0.15 M HClO4or 0.15 M Mg(ClO4)2in CH3CN. Linear plots of kobsversus substrate concentration are reported inFigures S4−S39.
■
DISCUSSIONThe data shown inTables 2and3indicate that the presence of the N-heteroaromatic ring in QINO results in a significant increase in HAT rate constants involving all the substrates. The higher reactivity of QINO compared to that of PINO can be
rationalized on the basis of both enthalpic and polar effects. We found that the BDENO−H value for NHQI determined experimentally by the EPR radical equilibration technique is ∼0.7 kcal/mol higher than that of NHPI and 1.1 kcal/mol
Figure 7.Optimized structures for the pre-reaction, transition state, and postreaction structures for the reaction of (a) QINO and (b) PINO with toluene. Structures were obtained using the B3LYP-DCP/6-31+G(2d,2p) approach with the SMD solvation (acetonitrile) model. Key distances shown are in angstroms. Key: carbon, gray; hydrogen, white; oxygen, red; nitrogen, blue. The geometric centers of thefive- and six-membered rings are shown as purple spheres.
Figure 8. Dependence of the observed rate constants (kobs) on
substrate concentration for the reactions of QINO with alkylaromatics measured in a CH3CN solution at 25°C by following the decay of
QINO at 390 nm.
Table 2. Second-Order Rate Constants (kH) for Hydrogen Atom Transfer (HAT) from Alkylaromatics to QINO and PINO Measured in CH3CN at 25°C
aAverage of at least three independent determinations. Error of±5%.
Statistically corrected for the number of labile hydrogens. Correlation coefficients: 0.982 < r2< 0.999.bIn the presence of 0.15 M HClO
4.cIn
the presence of 0.15 M Mg(ClO4)2.dSee ref12.eIn the presence of
0.1 M HClO4.
higher as determined by the B3LYP-DCP approach in solvent. The introduction of an electron-withdrawing nitrogen atom into the aromatic ring of NHQI results in a destabilization of the charge-separated resonance structure of the N-oxyl radical (seeFigure 2),2cthereby increasing the BDENO−H.
The reactivity of N-oxyl radicals in the HAT processes is determined by both the enthalpic requirements of the C−H and NO−H bond dissociation energies and by the polar effects that may stabilize the activated complex.6 The introduction of an N atom into the aromatic ring of QINO leads to a stabilization of the activated complex by increasing the electron density on QINO. Figure 7 shows an example of these stabilizing effects in the TS for the HAT from substituted toluenes.
The computational modeling of the abstraction of the benzylic hydrogen from toluene by QINO and PINO provides additional insights into the role of charge transfer stabilization of TS structures. The hydrogen transfer occurs by the same mechanism described in the previous study on the reactions of activated phenols with PINO and other N-oxyl radicals.6g,8The pre-reaction complex that is formed between the reactants has a cisoid structure that is stabilized throughπ-stacking interactions (see Figure 7). Relative to the separated reactants, the complexes have free energies of 5.5 kcal/mol for QINO and 4.6 kcal/mol for PINO. The cisoid arrangements are maintained in the TS structures, allowing a charge transfer contribution to the HAT process through the π-overlap between the benzylic and QINO aromatic rings.31 The calculations support the expectation that the inclusion of a nitrogen atom in the aryl ring of the abstracting radical, as in QINO, allows for a greater degree of charge transfer in the TS.
Specifically, an additional 0.06 electron is transferred between the QINO and benzyl moieties in the TS structure than in the analogous TS structure involving PINO.
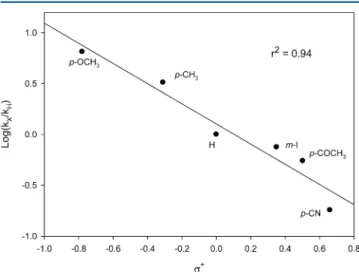
The kH values for the HAT from alkylaromatics to QINO reported in Table 2 show an increase in the following order: cumene > ethylbenzene > toluene. This is expected from the series of decreasing C−H BDEs from the tertiary C−H bond of cumene (BDE = 83.2± 1 kcal mol−1)32to the primary C−H bond in toluene (BDE = 89.7± 1.2 kcal mol−1)32and is in line with the HAT promoted by PINO in acetic acid2b and in CH3CN (seeTable 2). For the series of substituted toluenes, as expected for a HAT process promoted by electrophilic radicals and in accordance with the enthalpic and polar effects discussed above, the kH values increase with the electron donating strength of the aryl substituent. When the log(kHX/kHH) values for the reactions of QINO with substituted toluenes were plotted against the Okamoto−Brown substituent constants σ+
, a good Hammett-type correlations was obtained (Figure 9).33
The negativeρ value (−0.99),34as well as the better linearity obtained with the σ+ rather than the σ constants, is in accordance with the significant polar effects associated with the development of a partial positive charge on the benzylic position in the HAT TS.
Table 3shows the rate constants for the HAT from a series of hydrogen atom donors. Rate constants for the HAT from C−H bonds in unactivated aliphatic hydrocarbons (cyclo-hexane and adamantane), as expected, are significantly lower than those determined with alkylaromatics. The increase in reactivity observed for QINO as compared to that for PINO is of fundamental importance for these substrates that are characterized by relatively high C−H BDE values (99.5 kcal/ mol for cyclohexane and 96.2 and 98.4 kcal/mol for tertiary and secondary C−H bonds, respectively, in adamantane).32
The higher reactivity observed in HAT from alcohols, ethers (THF), and amides (DMF) is in line with the enthalpic and polar effects associated with electron rich C−H bonds in α position to heteroatoms, which can stabilize the partial positive charge that develops on carbon in the HAT.
The presence of the heteroaromatic nitrogen in NHQI allows a further activation of the corresponding N-oxyl radical in HAT processes by adding Brønsted or Lewis acids; in fact, Table 3. Second-Order Rate Constants (kH) for Hydrogen
Atom Transfer (HAT) from Aliphatic Hydrocarbons, Alcohols, Aldehydes, Ethers, and Amides to QINO and PINO Measured in CH3CN at 25°C
aAverage of at least three independent determinations. Error of±5%.
Correlation coefficients: 0.972 < r2< 0.999.bSee ref12.
Figure 9.Hammett plot for the reaction of substituted toluenes with QINO in CH3CN at 25°C.
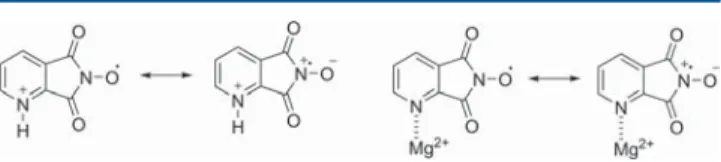
the protonation or coordination of the N atom decreases the electron density on QINO (Figure 10).35
An increased electrophilicity of QINO would result as a consequence of the destabilization of the charge-separated resonance structures shown in Figure 10. In this context, it should be mentioned that through complexation of NHQI with cobalt and copper ions it was possible to improve the activity of NHQI in the oxidation of toluene. This activating effect has been interpreted on the basis of an electron-withdrawing effect produced by the complexation of the N-heteroaromatic ring with the metal salts. The complexation of NHQI with CuCl2 has been also confirmed through real time in situ FTIR spectral analysis.10
To investigate the effect of acid additives on the QINO reactivity in HAT process, kinetic studies with alkylaromatics were also performed in the presence of 0.15 M HClO4and 0.15 M Mg(ClO4)2.
Considering the electron withdrawing effect of the two imidic carbonyls, the pKavalue for the pyridinium cation (12.3 in CH3CN)36 is much higher than that of HClO4 (2.0 in CH3CN);
36
thus, the protonation of QINO to give QINOH+• in 0.15 M HClO4should be almost quantitative. Accordingly, a variation of the UV−vis spectra of QINO, generated by oxidation of NHQI (1.0 mM) with CAN (0.5 mM) in CH3CN, was observed as a function of increasing the HClO4 concentration to a value of 0.15 M (seeFigure S40).
The data reported inTable 2indicate increases in reactivity of more than 1 order of magnitude, in the presence of 0.15 M HClO4, for all the alkylaromatics tested. This is consistent with thefindings based on the favorable enthalpic and polar effects associated with the protonation of QINO. It should be noted that a smaller increase in the kH value for the HAT from alkylaromatics [ethylbenzene and p-xylene (seeTable 2)] in the presence of HClO4was also observed for PINO in a previous study.12 The increase in reactivity with PINO has been attributed to the protonation of imidic carbonyl groups.
Compared to that with HClO4, a lower degree of activation was observed in the presence of 0.15 M MgClO4. For example, the kHvalue for HAT from toluene to QINO increased from 0.2 to 0.33 M−1 s−1 after addition of the Lewis acid. This behavior reflects a minor decrease in electron density in QINO compared to that of perchloric acid.
The increase in QINO electrophilicity caused by the protonation of the heteroaromatic nitrogen is in accordance with the more negative ρ value (−1.8) determined in the Hammett correlation (see Figure S42) for the HAT process from substituted toluenes to QINO observed in the presence of 0.15 M HClO4.
In conclusion, the results reported in this study clearly indicate that QINO has hydrogen abstracting ability that is greater than that of PINO in HAT processes from all the classes of organic compounds investigated. This supports the use of NHQI as a convenient substitute for NHPI in the catalytic aerobic oxidations of substrates such as aliphatic
hydrocarbons that have relatively high C−H bond dissociation energies.
■
EXPERIMENTAL SECTIONMaterials. Acetonitrile (HPLC grade), NHPI, (NH4)2Ce(NO3)6,
Mg(ClO4)2, and HClO4 were commercially available and used as
received. NHQI was synthesized by reaction of pyridine-2,3-dicarboxylic acid anhydride37 with hydroxylamine hydrochloride following a procedure reported in the literature.10
Toluene, ethylbenzene, cumene, p-xylene, p-methoxytoluene, m-iodotoluene, 4-methylacetophenone, p-tolunitrile, cyclohexane, ada-mantane, methanol, ethanol, 2-propanol, tetrahydrofuran, N,N-dimethylformamide, and p-chlorobenzaldehyde were commercially available at their highest purity and used as received.
Kinetic Studies. QINO and PINO were generated by oxidation of the corresponding N-hydroxyimides (NHQI and NHPI) (1 mM) with 0.4 mM cerium(IV) ammonium nitrate (CAN) in CH3CN at 25°C.
HAT reactions of QINO with alkylaromatics were investigated in the absence of acid additives or in the presence of HClO4(0.15 M) or
Mg(ClO4)2(0.15 M). After the generation of the N-oxyl radicals, an
excess of substrate was added to ensure the process occurred under pseudo-first-order conditions (final concentrations of 1.8−300 mM), and the absorbance change was monitored at 390 nm for QINO and 380 nm for PINO. For all the substrates investigated, each kinetic trace obeyedfirst-order kinetics. Second-order rate constants were obtained from the slopes of plots of the observed rate constants (kobs) versus
substrate concentration. Rate constants are reported as the average of at least three independent determinations with an error of±5%.
■
ASSOCIATED CONTENT*
S Supporting InformationThe Supporting Information is available free of charge on the
ACS Publications websiteat DOI:10.1021/acs.joc.7b00687. Instrumentation; determination of the O−H BDE values by EPR measurements; UV−vis spectroscopic character-ization of QINO; EPR spectrum of QINO obtained by oxidation of NHQI with cerium(IV) ammonium nitrate (CAN) in CH3CN at 25 °C; EPR spectrum of an irradiated CH3CN solution containing di-tert-butyl peroxide, NHQI, and 4-CH3OCO-NHPI; plots of kobs versus substrate concentration for the reactions of QINO and PINO with alkylaromatics, aliphatic hydrocarbons, alcohols, aldehydes, ethers, and amides; UV−vis spectra of QINO in MeCN and HClO4(0−0.2 M); Hammett plot for the HAT process from substituted toluenes to PINO in CH3CN and to QINO in CH3CN and 0.15 M HClO4; gas- and solvent-phase (acetonitrile) computa-tional results for HAT reaction of QINO and PINO with toluene; optimized structures of the transition states for the reaction of QINO and PINO with toluene; and Cartesian coordinates of the optimized structures of the reaction steps (PDF)
■
AUTHOR INFORMATION Corresponding Author *E-mail:osvaldo.lanzalunga@uniroma1.it. ORCID Osvaldo Lanzalunga: 0000-0002-0532-1888 Andrea Lapi:0000-0001-9728-8132 NotesThe authors declare no competingfinancial interest.
Figure 10.Resonance structures of QINO in the presence of Brønsted or Lewis acids.
■
ACKNOWLEDGMENTSThanks are due to the Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR), for financial support and to the CIRCC, Interuniversity Consortium of Chemical Catalysis and Reactivity. We thank Prof. M. Bietti and Dr. M. Salamone for the use of LFP equipment. G.A.D. thanks the National Sciences and Engineering Research Council of Canada, the Canadian Foundation for Innovation, and the University of British Columbia forfinancial support and Compute Canada for access to computational resources.
■
REFERENCES(1) (a) Ishii, Y.; Sakaguchi, S.; Iwahama, T. Adv. Synth. Catal. 2001, 343, 393−427. (b) Sheldon, R. A.; Arends, I. W. C. E. Adv. Synth. Catal. 2004, 346, 1051−1071. (c) Recupero, F.; Punta, C. Chem. Rev. 2007, 107, 3800−3842. (d) Coseri, S. Catal. Rev.: Sci. Eng. 2009, 51, 218−292. (e) Melone, L.; Punta, C. Beilstein J. Org. Chem. 2013, 9, 1296−1310. (f) Wertz, S.; Studer, A. Green Chem. 2013, 15, 3116− 3134. (g) Chen, K.; Zhang, P.; Wang, Y.; Li, H. Green Chem. 2014, 16, 2344−2374 and references cited therein.
(2) (a) Ueda, C.; Noyama, M.; Ohmori, H.; Masui, M. Chem. Pharm. Bull. 1987, 35, 1372−1377. (b) Koshino, N.; Cai, Y.; Espenson, J. H. J. Phys. Chem. A 2003, 107, 4262−4267. (c) Amorati, R.; Lucarini, M.; Mugnaini, V.; Pedulli, G. F.; Minisci, F.; Recupero, F.; Fontana, F.; Astolfi, P.; Greci, L. J. Org. Chem. 2003, 68, 1747−1754. (d) Koshino, N.; Saha, B.; Espenson, J. H. J. Org. Chem. 2003, 68, 9364−9370.
(3) (a) Galli, C.; Gentili, P.; Lanzalunga, O.; Lucarini, M.; Pedulli, G. F. Chem. Commun. 2004, 2356−2357. (b) Brandi, P.; Galli, C.; Gentili, P. J. Org. Chem. 2005, 70, 9521−9528. (c) Galli, C.; Gentili, P.; Lanzalunga, O. Angew. Chem., Int. Ed. 2008, 47, 4790−4796. (d) Coniglio, A.; Galli, C.; Gentili, P.; Vadalà, R. J. Mol. Catal. B: Enzym. 2008, 50, 40−49. (e) Coniglio, A.; Galli, C.; Gentili, P.; Vadalà, R. Org. Biomol. Chem. 2009, 7, 155−160.
(4) The initial value of 88.1 kcal mol−1has been recalculated using the corrected O−H BDE value for the reference compound BHT (ref
5).
(5) (a) Mulder, P.; Korth, H.-G.; Pratt, D. A.; DiLabio, G. A.; Valgimigli, L.; Pedulli, G. F.; Ingold, K. U. J. Phys. Chem. A 2005, 109, 2647−2655. (b) Capraro, M. G.; Franchi, P.; Lanzalunga, O.; Lapi, A.; Lucarini, M. J. Org. Chem. 2014, 79, 6435−6443.
(6) (a) Minisci, F.; Punta, C.; Recupero, F.; Fontana, F.; Pedulli, G. F. J. Org. Chem. 2002, 67, 2671−2676. (b) Minisci, F.; Punta, C.; Recupero, F.; Fontana, F.; Pedulli, G. F. Chem. Commun. 2002, 688− 689. (c) Cecchetto, A.; Minisci, F.; Recupero, F.; Fontana, F.; Pedulli, G. F. Tetrahedron Lett. 2002, 43, 3605−3607. (d) Minisci, F.; Recupero, F.; Cecchetto, A.; Gambarotti, C.; Punta, C.; Faletti, R.; Paganelli, R.; Pedulli, G. F. Eur. J. Org. Chem. 2004, 2004, 109−119. (e) Baciocchi, E.; Gerini, M. F.; Lanzalunga, O. J. Org. Chem. 2004, 69, 8963−8966. (f) Minisci, F.; Punta, C.; Recupero, F. J. Mol. Catal. A: Chem. 2006, 251, 129−149. (g) D’Alfonso, C.; Bietti, M.; DiLabio, G. A.; Lanzalunga, O.; Salamone, M. J. Org. Chem. 2013, 78, 1026−1037. (7) (a) Annunziatini, C.; Gerini, M. F.; Lanzalunga, O.; Lucarini, M. J. Org. Chem. 2004, 69, 3431−3438. (b) Cai, Y.; Koshino, N.; Saha, B.; Espenson, J. H. J. Org. Chem. 2005, 70, 238−243. (c) Annunziatini, C.; Baiocco, P.; Gerini, M. F.; Lanzalunga, O.; Sjogren, B. J. Mol. Catal. B: Enzym. 2005, 32, 89−96. (d) Sun, Y.; Zhang, W.; Hu, X.; Li, H. J. Phys. Chem. B 2010, 114, 4862−4869. (e) D’Alfonso, C.; Lanzalunga, A.; Lapi, A.; Vadalà, R. Tetrahedron 2014, 70, 3049−3055. (f) Chen, K.; Yao, J.; Chen, Z.; Li, H. J. Catal. 2015, 331, 76−85.
(8) Mazzonna, M.; Bietti, M.; DiLabio, G. A.; Lanzalunga, O.; Salamone, M. J. Org. Chem. 2014, 79, 5209−5218.
(9) (a) Wentzel, B. B.; Donners, M. P. J.; Alsters, P. L.; Feiters, M. C.; Nolte, R. J. M. Tetrahedron 2000, 56, 7797−7803. (b) Zhang, Q.; Chen, H.; Ma, H.; Miao, H.; Zhang, Z.; Sun, Z.; Xu, J. J. Chem. Technol. Biotechnol. 2008, 83, 1364−1369.
(10) Zhang, Q.; Chen, C.; Xu, J.; Wang, F.; Gao, J.; Xia, C. Adv. Synth. Catal. 2011, 353, 226−230.
(11) Zhao, Q.; Chen, K.; Zhang, W.; Yao, Y.; Li, H. J. Mol. Catal. A: Chem. 2015, 402, 79−82.
(12) Bietti, M.; Forcina, V.; Lanzalunga, O.; Lapi, A.; Martin, T.; Mazzonna, M.; Salamone, M. J. Org. Chem. 2016, 81, 11924−11931.
(13) Avila, D. V.; Brown, C. E.; Ingold, K. U.; Lusztyk, J. J. Am. Chem. Soc. 1993, 115, 466−470.
(14) (a) Avila, D. V.; Lusztyk, J.; Ingold, K. U. J. Am. Chem. Soc. 1992, 114, 6576−6577. (b) Avila, D. V.; Ingold, K. U.; Di Nardo, A. A.; Zerbetto, F.; Zgierski, M. Z.; Lusztyk, J. J. Am. Chem. Soc. 1995, 117, 2711−2718.
(15) (a) Coseri, S.; Mendenhall, G. D.; Ingold, K. U. J. Org. Chem. 2005, 70, 4629−4636. (b) Baciocchi, E.; Bietti, M.; Gerini, M. F.; Lanzalunga, O. J. Org. Chem. 2005, 70, 5144−5149. (c) Baciocchi, E.; Bietti, M.; Di Fusco, M.; Lanzalunga. J. Org. Chem. 2007, 72, 8748− 8754.
(16) The aNvalue of QINO is higher than that previously reported
for PINO (aN= 4.76), in accordance with the electron withdrawing
effect of the QINO N-heteroaromatic ring (see ref7a).
(17) Da Silva, G.; Bozzelli, J. W. J. Phys. Chem. C 2007, 111, 5760− 5765.
(18) The value of 88.9 kcal mol−1 reported in ref 7a has been recalculated using the corrected O−H BDE value for the reference compound BHT (ref5).
(19) Franchi, P.; Mezzina, E.; Lucarini, M. J. Am. Chem. Soc. 2014, 136, 1250−1252.
(20) The error in the BDE measured by EPR (±0.6 kcal/mol) is related to the error reported for the original BDE measurement of NHPI used as reference (ref2c). However,ΔBDE errors in the EPR measurements are generally smaller (<0.3 kcal/mol). The accuracy of the EPR method relies on the fact that even relatively large errors in the measurement of radical concentrations and therefore of the equilibrium constant, Keq, give rise to small errors in the ΔBDEs
because of the logarithmic relation connecting these two quantities. (21) (a) Becke, A. D. J. Chem. Phys. 1993, 98, 5648−5652. (b) Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785−789.
(22) (a) Torres, E.; DiLabio, G. A. J. Phys. Chem. Lett. 2012, 3, 1738−1744. (b) DiLabio, G. A.; Koleini, M.; Torres, E. Theor. Chem. Acc. 2013, 132, 1389.
(23) Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378−6396.
(24) Montgomery, J. A., Jr.; Frisch, M. J.; Ochterski, J. W.; Petersson, G. A. J. Chem. Phys. 2000, 112, 6532−6542.
(25) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; et al. Gaussian 09, revision D.01; Gaussian, Inc.: Wallingford, CT, 2009.
(26) The computed gas-phase BDEs are lower than the experimental BDEs measured in polar solvents due to the hydrogen bond of the N-hydroxylamines with the solvent (see ref8).
(27) DiLabio, G. A.; Johnson, E. R. J. Am. Chem. Soc. 2007, 129, 6199−6203.
(28) Harshan, A. K.; Yu, T.; Soudackov, A. V.; Hammes-Schiffer, S. J. Am. Chem. Soc. 2015, 137, 13545−13555.
(29) The use of the LFP technique for the generation and reactivity studies of short-lived aminoxyl radicals is limited to very fast reaction processes [on the micro- and millisecond time scales (see refs8,13b, and13c)], much faster than those reported in the study presented here.
(30) The pseudo-first-order rate constant for the decay of QINO is described by the equation kobs= kd+ kH[Ar-R], where kdis the QINO
self-decay rate constant. Thus, in the plot of kobsversus [Ar-R], the
intercept on the kobsaxis ([Ar-R] = 0) should not be on the origin but
should be equal to kd. Because the extrapolated intercept value is
always subjected to significant errors, a precise kddetermination using
this approach is not allowed.
(31) It is rather difficult to determine with accuracy the contribution of charge transfer fromπ-stacking between the aryl rings of PINO/ QINO and aromatic substrates to the HAT process because it is
summed to the enthalpic and other polar effects, also present in the non-aryl hydrocarbons. On these bases, a direct comparison of the relative weights of these effects with different substrates is not possible. (32) Luo, Y.-R. Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, 2007.
(33) A similar behavior in Hammett-type correlations has been reported for hydrogen abstraction promoted by other electrophilic radicals: Pryor, W. A.; Lin, T. H.; Stanley, J. P.; Henderson, R. W. J. Am. Chem. Soc. 1973, 95, 6993. Walling, C.; Jacknow, B. B. J. Am. Chem. Soc. 1960, 82, 6113. Gilliom, R. D.; Ward, B. F., Jr. J. Am. Chem. Soc. 1965, 87, 3944. Kennedy, B. R.; Ingold, K. U. Can. J. Chem. 1966, 44, 2381. Russell, G. A.; Williamson, R. C., Jr. J. Am. Chem. Soc. 1964, 86, 2357. Pearson, R. E.; Martin, J. C. J. Am. Chem. Soc. 1963, 85, 354. Huyser, E. S. J. Am. Chem. Soc. 1960, 82, 394.
(34) Theρ value is in line with those determined for the reactions of substituted toluenes with PINO in AcOH (−1.3) (ref 2d) and in CH3CN [−1.0 (seeFigure S41)].
(35) The good hydrogen abstracting ability of QINOH+•, generated from acid−base reaction of NHQI and 4-carboxyl-N-hydroxyphthali-mide, was taken into account to explain the high catalytic activity in the aerobic oxidation of ethylbenzene (ref11).
(36) Izutsu, K. Acid-base Dissociation Constant in Dipolar Aprotic Solvents; Alden Press: Oxford, U.K., 1990.
(37) Gould, K. J.; Hacker, N. P.; McOmie, J. F. W.; Perry, D. H. J. Chem. Soc., Perkin Trans. 1 1980, 1834−1840.