Directed Evolution in Human Cells via Adenoviral Replication by
Chet Michael Berman
B.S. in Biochemistry, BA in Chemistry (2012) Brandeis University, Waltham, MA
Submitted to the Department of Chemistry
In Partial Fulfillment of the Requirements for the Degree of
Doctor of Philosophy in Chemistry At the
Massachusetts Institute of Technology February, 2019
C 2019 Massachusetts Institute of Technology All rights reserved
Signature redacted
Signature of the Author:Department of Che try, September 21, 2018
Signature redacted
Certified by:
Matthew D. Shou Whitehead Career Development Associate Professor
Thesis supervisor
Signature redacted
Accepted by:MASSACHUSETTS INSTITUTE Robert W. Field
OF TECHNOLOGY Chairman, Department Committee on Graduate Students
MAR2Z1
.2]
This doctoral thesis has been examined by a committee of the Department of Chemistry as follows:
Signature redacted
John M. Essigmann William R. (1956) & Betsy P. Leitch Professor in Residence Professor of Chemistry, Toxicology, and Biological Engineering Director, MIT Center for Environmental Health Sciences (CEHS) Department of Chemistry, and Department of Biological Engineering, MIT Thesis committee chair
Signature
redacted-Matthew D. Shoulders Whitehead Career Development Associate Professor Associate Member, Broad Institute at Harvard and MIT Investigator, Center for Skeletal Research at Massachusetts General Hospital Member, MIT Center for Environmental Health Sciences Department of Chemistry, MIT Thesis supervisor
Signature redacted
K. Dane Wittrup Carbon P. Dubbs Professor in Chemical Engineering and Biological Engineering Associate Director for Engineering, Koch Institute for Integrative Cancer Research Department of Biological Engineering, and Department of Chemical Engineering, MIT Thesis Committee Member
Directed Evolution in Human Cells via Adenoviral Replication by
Chet Michael Berman
Submitted to the Department of Chemistry On September 28", 2018 in Partial Fulfillment of the
Requirements for the Degree of Doctor of Philosophy in Biological Chemistry Abstract
The discovery and optimization of biomolecules that reliably function in metazoan cells is imperative for both basic biological research, and therapeutic development. Typically, researchers turn to directed evolution either in vitro, in bacteria, or in yeast, to mutate, select, and amplify biomolecules of interest (BOls) with new and highly-tailored activities. Unfortunately, BOIs evolved in these environments often fail to function when translated into the complex environment of metazoan cells. Unique metazoan biology such as sophisticated proteostasis networks, complex cell signaling pathways, distinctive post-translational modifications, cellular trafficking, and highly regulated chromatin architecture can all derail the activity of BOIs evolved in simpler systems. Current approaches to directed evolution in metazoan cells rely on labor-intensive and time-consuming screening approaches that have a high potential for false positives. Robust approaches for directed evolution directly in human cells are profoundly needed.
In this thesis, I describe the development, characterization, and application of a broadly applicable platform for directed evolution of diverse BOIs directly in human cells. The platform relies on a partially gutted adenovirus lacking multiple genes, including the essential DNA polymerase and protease genes, features that allow us to evolve BOIs encoded by genes as large as 7 kb while attaining the mutation rates and enforcing the selection pressure required for successful directed evolution of complex function. High mutagenesis rates are attained by trans-complementation of an engineered, highly error-prone form of the adenoviral polymerase. Selection pressure that couples desired BOI functions to adenoviral propagation is achieved by linking the functionality of the encoded BOI to the production of adenoviral protease activity by the human cell. This platform makes it possible, in principle, to evolve any biomolecule activity that can be coupled to protease expression or activation by serially passaging adenovirus carrying the BOI. As proof-of-concept, we use the platform to evolve, directly in the human cell environment, several transcription factor variants that maintain high levels of function while gaining resistance to a small molecule inhibitor. We anticipate that this platform will substantially expand the repertoire of biomolecules that can be reliably and robustly engineered for both research and therapeutic applications in metazoan systems.
Thesis Supervisor: Matthew D. Shoulders
Directed Evolution in Human Cells via Adenoviral
Replication
Sometimes the biggest challenges end up being the best things that happen in your life.
Acknowledgements
I first want to thank my thesis advisor, Prof. Matthew Shoulders. Matt has been incredibly supportive and helpful throughout my time at MIT. When I first joined his lab, I knew nothing about how to do mammalian cell biology. Matt made sure I got caught up quickly, and always made time to meet with me to make sure I was adjusting properly. He also helped me extensively edit my fellowship applications, and was instrumental in helping me secure funding through the NIH Biotechnology Training Program. He constantly pushed me to be a better writer, and a better scientist. Through his mentorship, I learned how to think scientifically about questions, and generate new experimental plans to address my hypotheses. He was always available to lend a helping hand. Even at his busiest, I could always count on him to meet with me about whatever concern I had, be it science-related, or life-related. He was incredibly understanding with giving me the time I needed to plan my wedding, and with my move to New Hampshire towards the end of my PhD. I could not have asked for a better thesis advisor, and would not be here today without his mentorship and support.
I would also like to thank my committee members, Prof. John Essigmann, and Prof. K. Dane Wittrup. During our annual meetings, John would always ask me thought-provoking questions that led me to think about my project and my work in a new light. His perspective was incredibly valuable, and I am thankful for his input. While I only formally spoke to Dane once about my work, it came at a pivotal point in the project. His experimental input transformed how I approached my project experimentally, and he helped me understand how to better think about directed evolution. I would also like to thank my former thesis committee chair Prof. Alice Ting. Her valuable advice in early meetings helped me think more deeply about my project, and helped me approach scientific questions with more rigor. I also want to thank my undergraduate advisor, Prof. Gregory Petsko for taking me on as a freshman in his lab, and supporting my research throughout college. He also helped me find a research position after college, and wrote me letters of recommendation for graduate school and for fellowships.
I would like to thank our collaborators at Leiden University Medical Center, Prof Robert Hoeben and Taco Uil (now at Jenssen Pharmaceuticals). Taco and Rob's work on the error-prone adenoviral polymerase was instrumental to the success of our project, and their willingness to work with us as we prepared our manuscript for submission was invaluable. Rob, in particular has been incredibly supportive throughout my time at MIT. Whenever we were unsure how to proceed with our project, we would call Rob, and he would usually come up with several new directions on how we should proceed. He was always gracious with his time, and his hospitality when we visited his group in Leiden was deeply appreciated.
None of the work I conducted would have been possible without the outstanding core facility staff at MIT. The BMC staff was always available to help if I had trouble with the lightcyclers, and they were swift, thorough, and professional in running the MiSeq for my sequencing experiments. In particular, I want to thank Vincent Butty for his help in analyzing our sequencing data, and for helping me better understand how to think about large sequencing data sets. Stuart Levine was a terrific source of knowledge, and helped us make sure we were asking the right scientific questions, and preparing the correct experiments. I also want to thank Michael Jennings, Michele Griffin, and Glenn Paradis at the flow cytometry core facility. With the hundreds of flow cytometry experiments I ran while at MIT, they were always there to help me if I ran into any problems. Jaime Cheah and Christian Soule helped us run the high-throughput screening experiments. Their expertise made my life significantly easier. They were also a lot of fun, and I would
look for excuses to go to the HTS core facility to talk football with them.
I want to extend a special thank you to my partners in crime, Louis Papa and Sam Hendel. Both Sam and Louis are amazing people, and have always been willing to help me with whatever problem I had. I could always count on them to help me run experiments while I was away, or lighten the load if I was overwhelmed. Louis has been my close colleague on this project for four of five years, and was an
instrumental piece of nearly every facet of the project. His tireless effort figuring out the colossal recombineering problems we ran into was both impressive and extremely important for enabling almost every experiment we ran. Louis is also a terrific friend and colleague. I had a great time traveling with him to the Netherlands and New Orleans, and regret not having the chance to ski with him so I could show him a thing or two. Sam joined the lab, and the project, at a really critical time. His nearly boundless enthusiasm provided the jump-start we needed to get our experiments working properly. His work getting adenoviral protease trans-complementation, and running directed evolution was extremely impressive. Sam was a great colleague, and I always enjoy talking to him in the morning and during lunch. I cannot be more thankful to Sam and Lou for everything they have done for the project, and for me.
I want to thank all the members of the Shoulders Lab for an incredible five years. The Shoulders lab environment is friendly, helpful, industrious, efficient, and supportive. I want to extend a special thank you specifically to Chris Moore for all his hard work on this project, and for hosting an awesome lab barbecue with amazing ribs. I want to thank Duc, Azade and Rebecca for keeping me company in the far corner of the lab, and for keeping me humble. I want to especially thank Duc for his assistance in synthesizing the adenoviral protease inhibitor. I want to thank Andrew for teaching me how to science properly, and dealing with my persistent and endless questions during my first couple of years and beyond. I want to thank Chris Richardson for proofreading many of the documents I prepped over the years, and for always being willing to share his "interesting" opinions. I also want to thank Madeline and Angela for being an awesome cohort, and keeping me company first thing in the morning. Special thanks to Angela for being a fun neighbor for four years before abandoning me, and for being a terrific friend.
I would like to thank the ChemREFS for teaching me important interpersonal skills such as active listening and conflict resolution. These skills have been immensely helpful both personally and professionally.
Life would be considerably less enjoyable were it not for the tremendous group of friends I made in graduate school and beyond. I could always count on Alex Mijalis to keep me company any time I wanted, whether we were failing to convince one another to go to the gym, or bouncing experiment ideas back and forth. Yisu Han and Kenny Kang were also terrific friends throughout graduate school. I want to specifically thank Kenny for helping me move on Allston Christmas. That is a debt that can never be repaid. I want to thank Seth Lieblich for taking me on in the lab in undergrad, and being a terrific source of wisdom. Finally, I want to thank my friends from college and high school, including David Dawson, Julius Johnson, Clark Soucy, Brian Goldman, and Michael Rosenfeld. Your continued support and friendship is really appreciated.
I would not have gotten to where I am today were it not for the amazing support I received from my family. I cannot thank my Mom and Dad enough for everything they have done for me. They have always supported me no matter what I wanted to do, and pushed me to reach my goals. My mother-in-law and father-in-law have been a second set of parents to me, and I always feel at home when I come visit. My brother and sister Scott and Dani are both amazing, and my absolute best friends in the world. My brother-in-law is always fun to hang out with, and I always love seeing him when I come visit. Nana, Papa, Lori, Steve, Noah, and Josh are always willing to go out to dinner with me, and I love seeing them any chance I get. They have been incredibly supportive for the nearly ten years I have lived in the Boston area, and I cannot thank them enough for everything they have done for me.
Finally, I want to thank my kind, caring, loving, smart, funny, all-around amazing wife Lauren. I know that no matter how stressful work gets, the second I see her, I am at ease. Even though we spend virtually every waking moment that we're not working together, I never tire of her company. She supports me in more ways than I can count. If I am feeling down or anxious, she knows exactly what to say to make me feel better. If I need her help with something, she always does it with a smile. Most importantly, she
cooks me delicious meals, and I therefore eat like a king. I could not have asked for a better partner to live my life with.
I want to thank the NIH Biotechnology Training Program for funding three years of my PhD, and Rubius Therapeutics and Tiffany Chen for the summer internship opportunity.
I want to extend a special thanks to Matt, Sam, Louis, Angela, and Lauren for their helpful comments and suggestions in preparing my thesis.
Table of Contents
Abstract ... 3
Acknow ledgem ents ... 6
Table of Contents ... 9
List of Figures ... 13
List of Tables ... 15
Abbreviations ... 16
Chapter 1: Directed Evolution Strategies for Function in Metazoan Systems...19
1. 1. In tro d u ctio n ... 2 0 1.2. In vitro directed evolution ... 22
1.2.1. In vitro m utagenesis ... 22
1.2.2. In vitro screening and selection for functional B01s... 23
1.2.3. In vitro directed evolution of BOIs for function in metazoan cells... 25
1.3. Directed evolution in bacteria and yeast ... 26
1.3.1. Directed evolution in bacteria for function in metazoan cells... 27
1.3.2. Directed evolution in yeast for function in metazoan cells ... 30
1.4. Directed evolution in metazoan systems...3 1 1 .4.1. M utagenesis in metazoan system s... 32
1.4.2. Screening and selection in metazoan system s... 36
1.4.3. Directed evolution in human cells by viral evolution ... 38
1.4.4. Limitations of current approaches for directed evolution in metazoan cells...40
1.5. N ew approaches for continuous directed evolution in metazoan system s ... 41
1.6. References ... 43
Chapter 2: Design, Characterization, and Optimization of Components for Adenovirus-Mediated Directed Evolution in H um an Cells... 51
2. A. Author contributions ... 52
2.2. Introduction ... 53
2 .3 . R e su lts ... 5 7 2.3.1. Mutagenesis system and adenoviral DNA polymerase trans-complementation...57
2.3.2. Development of trans-complementation systems for putative selection schemes...64
2.4. Discussion ... 72
2.5. 1. Vectors and cloning ... 77
2.5.2. Cell Culture and Lentivirus Transduction ... 81
2.5.3. Determ ination of the m utagenic potential of EP-Pol ... 81
2.5.4. Im m unoblotting...82
2.5.5. RT-qPCR ... 82
2.5.6. Generating adenovirus from transfection ... 82
2.5.7. Trans-com plem entation assays ... 83
2.6. Acknow ledgem ents...86
2.7. References...87
Chapter 3: Adenovirus-Mediated Directed Evolution in Human Cells ... 93
3. 1. A uthor contributions ... 94
3.2. Introduction ... 95
3.3. Results ... 99
3.3.1. Im proving production of AdProt APo .adenoviruses... 99
3.3.2. Preparation of com ponents for directed evolution of tTA ... 99
3.3.3. Enrichm ent of active BOI variants in a tTA transcriptional circuit... 100
3.3.4. Expanding the dynamic range of selection using a selective adenoviral protease inhibitor 102 3.3.5. Proof-of concept directed evolution of functional tTA variants that acquire doxycycline resistance 104 3.3.6. Generalizable testing of selection circuits... 107
3.4. Discussion ... 112
3.5. M aterials and M ethods... 116
3.5.1. Vectors and cloning:... 116
3.5.2. Cell Culture and Lentivirus Transduction:... 120
3.5.3. RT-qPCR: ... 121
3.5.4. Generating adenovirus from transfection:... 121
3.5.5. Determ ining adenoviral titer by flow cytometry:... 122
3.5.6. Com petition experim ents: ... 122
3.5.7. Doxycycline dose response: ... 122
3.5.8. AdProt inhibitor experim ents:... 122
3.5.9. Continuous evolution workflow :... 123
3.5. 10. Analyzing prom oter activity in passaged viral supernatant: ... 124
3.5.11 .tTA evolution sequencing:... 124
3.5.12. Reverse genetics of tTA variants:... 124
3.5.14. Selection circuit experiments: ... 125
3.6. Acknowledgements ... 129
3 .7 . R eferen ces ... 13 0 Chapter 4: Improvements and Next-Generation Targets for Adenovirus-Mediated Directed Evolution in Hum an Cells...135
4 .1. Intro d uctio n ... 13 6 4.2. Improvements to adenovirus-mediated directed evolution in human cells ... 138
4.2.1. Effects of promoter strength on selection and adenovirus infectivity... 138
4.2.2. Targeted mutagenesis to increase the mutation rate without sacrificing adenovirus viability 139 4.2.3. Library size expansion using suspension cells ... 141
4.2.4. Negative selection for directed evolution of selectivity and specificity... 142
4.3. Next-generation targets for directed evolution in human cells ... 145
4.3.1. tRNA amino-acyl synthetase pairs ... 146
4.3.2. Zinc-Finger Nucleases for Genome Editing... 149
4.3.3. Analyzing the influence of proteostasis on the genesis of cancer... 150
4 .4 . S u m m ary ... 154
4 .5 . R efe re n ces ... 1 5 5 Appendix: Fluorescence Screening and Antibiotic Selection for Adenovirus-Mediated Directed Evolution...160
A . In tro d uctio n ... 16 1 A .2 R e su lts ... 1 6 1 A.2.1 FACS-based screening of positive BOI variants for adenovirus-mediated directed evolution ... 161
164 A.2.2 Antibiotic selection of positive BOI variants for adenovirus-mediated directed evolution ... 164
A.3 M aterials and M ethods ... 169
A.3.1 Vectors and cloning: ... 169
A.3.2 Cell Culture and Lentivirus Transduction: ... 172
A.3.3 Generating adenovirus from transfection: ... 172
A.3.4 Determining adenoviral titer by flow cytometry:... 173
A.3.5 Testing the eGFP reporter cell line by transient transfection of tTA: ... 173
A.3.6 Enrichment of active BOI variants via FACS: ... 173
A.3.7 Resazurin assay: ... 174
A.3.8 Time course of antibiotic inhibition of adenoviral replication: ... 174
A .3.1 0 Enrichm ent of tTA-expressing adenovirus: ... 175 A .4 References ... 177
List of Figures
Chapter 1: Directed Evolution Strategies for Function in Metazoan Systems
Figure 1.1 . D irected Evolution Cycle ... 21
Figure 1 .2 Strategies for in vitro m utagenesis... 24
Figure 1.3. In vitro selection by protein display... 25
Figure 1.4 Targeted, processive in vivo m utagenesis ... 28
Figure 1.5. Phage-assisted continuous evolution (PACE)... 29
F igure 1.6. Y east surface display ... 3 1 Figure 1.7. Mutagenesis through somatic hypermutation... 33
Figure 1.8. Targeted somatic hypermutation via RNA-guided nucleases...35
Figure 1.9. Fluorescent screening via robotic cell picking ... 38
Figure 1.10. Virus-based directed evolution in human cells ... 40
Chapter 2: Design, Characterization, and Optimization of Components for Adenovirus Mediated Directed Evolution in Human Cells Figure 2.1. Adenovirus-mediated directed evolution in human cells... 55
Figure 2.2. Locations of mutations in AdPol for increased mutagenesis... 58
Figure 2.3. Recombineering counterselection scheme to make a targeted, seamless deletion in the adenov iral geno m e ... 60
Figure 2.4. Trans-complementation of adenoviral polymerase ... 62
Figure 2.5. M utation R ate of EP-Pol...63
Figure 2.6. mRNA expression of E2A in AdPol-expressing cells... 66
Figure 2.7. pVI single colony expression analyzed by Western blot ... 68
Figure 2.8. Fiber domain architecture and cell line expression ... 69
Figure 2.9. mRNA expression of AProt in AdPol-expressing cells ... 70
Figure 2. 10. AdProt/AdPol adenovirus trans-complementation... 71
Chapter 3: Adenovirus-mediated Directed Evolution in Human Cells Figure 3.1. Summary of adenovirus-mediated, human cell-based directed evolution workflow ... 96
Figure 3.2. qPCR data of AdProt-expressing cell lines... 100
Figure 3.3. Demonstration of enrichment of a model biomolecule of interest ... 101
Figure 3.5. A denovirus protease inhibitor... 103
Figure 3.6 Directed evolution of doxycycline resistance in tTA... 105
Figure 3.7. Deep sequencing of evolved viral populations... 106
Figure 3.8. Reverse genetics to validate observed dox-resistant and CMV promoter mutants... 107
Figure 3.9. Generalized experiment to test diverse genetic selection circuits ... 108
Figure 3.10. Genetic selection circuits to evolve diverse functions... 109
Figure 3.11 Testing diverse selection circuits ... 111
Chapter 4: Improvements and Next Generation Targets for Adenovirus-Mediated Directed Evolution in Human Cells Figure 4.1. Adenovirus-mediated directed evolution in human cells... 137
Figure 4.2. Improvements to adenovirus-mediated directed evolution... 140
Figure 4.3. Possible negative selection strategies to prevent the evolution of nonspecific a ctiv itie s ... 14 4 Figure 4.4. Next generation directed evolution experiments ... 148
Figure 4.5. Studying the effects of the human proteostasis network on the evolution of o nco gen ic p roteins ... 153
Appendix: Fluorescent Screening and Antibiotic Selectionfor Adenovirus-Mediated Directed Evolution Figure A. 1. Screening positive BOI variants using an eGFP reporter gene ... 162
Figure A.2. tTA-induced eGFP expression in the reporter cell line ... 163
Figure A.3. Enrichment of adenoviruses encoding positive BOI variants using FACS... 164
Figure A.4. Resazurin assay to assess acute toxicity of common tissue culture antibiotics ... 165
Figure A.5. Time-course of antibiotic-mediated inhibition of adenoviral infection ... 166
Figure A.6. tTA-induced resistance to antibiotic mediated inhibition of adenoviral infection ... 167 Figure A.7. Enrichment of adenoviruses encoding positive BOI variants by antibiotic selection
List of Tables
Chapter 2: Design, Characterization, and Optimization of Components for Adenovirus Mediated Directed Evolution in Human Cells
Table 2.1. Adenoviruses constructed and used in this study ... 61
Table 2.2. C ell lines used in this study... 61
Table 2.3. M utation rate of EP-Pol ... 64
Table 2.4. Modifications to adenoviral vectors...83
Table 2.5. Prim ers used in this study ... 83
Chapter 3: Adenovirus-mediated Directed Evolution in Human Cells Table 3.1. A denoviruses used in this study ... 125
Table 3.2. C ell lines used in this study ... 126
T ab le 3 .3 . Prim ers em p loyed ... 126
Table 3.4. Modifications made to generate adenoviruses used in this study... 128
Appendix: Fluorescent Screening and Antibiotic Selection for Adenovirus-Mediated Directed Evolution Table A .1. A denoviruses used in this study... 175
Table A .2. C ell lines used in this study ... 1 75 Table A .3. Prim ers used in this study ... 175
Abbreviations
A genetic deletion
A549 an adenocarcinomic epithelial cell line aaRS amino-acyl tRNA synthetase
AdPol adenoviral DNA polymerase AdProt adenoviral protease
APOBEC apolipoprotein B mRNA editing enzyme, catalytic polypeptide like blastR blasticidin-S-deaminase
BOI biomolecule of Interest
BAC bacterial artificial chromosome
C Celsius
CAR coxsackie and adenovirus receptor CFP cyan fluorescent protein
CMV cytomegalovirus immediate-early promoter/enhancer
CPE cytopathic effect
cm centimeter
bp base pair
ccdB/ccdA control of cell death B/A
DAPI diamidino-2-phenylindole
DBD DNA-binding domain
dCas9 deactivated Cas9
DD destabilized domain
DNA deoxyribonucleic acid
DNase deoxyribonuclease
dNTP deoxynucleotide triphosphate
Dox doxycycline
E2A adenovirus early gene 2A E. coli Escherichia coli
eCFP enhanced cyan fluorescent protein eGFP enhanced green fluorescent protein eIF4F eukaryotic initiation factor 4F
EP-Pol error-prone adenoviral DNA polymerase FAB fragment antigen binding
FACS fluorescence-activated cell sorting
FKBP FK506 binding protein
FRB FKBP 1 2-rapamycin-binding domain
galK galactokinase gene
gRNA guide RNA
h hour
HA hemagglutinin epitope
HEK293 human embryonic kidney 293 cells
HEK293-F freestyle human embryonic kidney 293 cells HIV human immunodeficiency virus
hpi hours post-infection
Ig immunoglobulin
IU infectious units
Kan kanamycin resistance gene or kanamycin (depending on context)
Kb kilobase
LeuRS leucine amino-acyl tRNA synthetase
LOD limit of detection
tL microliter ptm micrometer jIM micromolar M molar mL milliliter mm millimeter mM millimolar min minutes
MOI multiplicity of infection
Muta-T7 rAPOBECI-T7 RNAP chimera
nCas9 nickase Cas9
nM nanomolar
nt nucleotide
oligo oligonucleotide
ORF open reading frame
PACE phage-assisted continuous evolution PCR polymerase chain reaction
PEI polyethylenimine
PPI protein-protein interaction puroR puromycin N-acetyl-transferase pVI adenovirus precursor protein VI
RNA ribonucleic acid
RNAi RNA interference
RRE rev response elements rtTA reverse tet-transactivator S. cerevisiae Saccharomyces cerevisiae
scFv single-chain variable fragments
SHM somatic hypermutation
SSA single-strand annealing system T7 RNAP T7 RNA polymerase
TAD transcriptional activation domain TALEN transcription activator-like effector nuclease TNFac tumor Necrosis Factor cc
TPL tripartite Leader
tetO tet operon
tetR tet repressor
TRE tet-responsive element
tRNA transfer RNA
tRNA Leu Leucine tRNA
tTA tet-transactivator protein (a.k.a. tet-OFF)
tTAwt tTA that binds to the wild-type operator sequence tTAmnut tTA that binds to the mutant operator sequence UAA unnatural amino acid
UGI uracil-DNA glycosylase VP48 3xVP16 transactivation domain VSV vesicular stomatitis virus
VSV-G vesicular stomatitis virus envelope ZFN zinc-finger nuclease
Chapter 1: Directed Evolution Strategies for Function in
Metazoan Systems
1.1. Introduction
The world as we know it would not exist without evolution. Biological evolution and natural selection have generated stunning diversity across species, through solely random mutational variation. Specifically, when an organism's genetic material is improperly replicated, while beneficial mutations sometimes arise, most often mutations are actually disadvantageous. Considering the fierce competition for scarce resources and harsh environments, typically the genetic traits that get selected, and those that survive moving forward, are those that impart an organism with an advantage for reproduction and/or long-term survival.
While advantageous genetic traits are more frequently amplified in natural biological evolution, humans have gone one step further and have co-opted the idea of natural selection to produce organisms with highly tailored traits through strategies such as selective breeding and artificial selection. Through artificial selection, humans have generated incredible species such as crops with enhanced caloric content and livestock optimized to plow our fields and sustain us with meat. At its core, artificial selection extends down to the molecular level, which is an arena where scientists can perform highly targeted selections on individual genes to select for enhanced traits. This process, termed molecular directed evolution, has enabled scientists to produce, among countless other functions, enzymes with new activities and fluorescent proteins with altered emission spectra.'
The process of directed evolution closely mimics biological evolution by natural selection (Figure 1.1). A gene of interest is mutated to generate a genetic library. The library is then expressed to form a library of biomolecules of interest (BOIs). Individual library members are selected for enhancement in the desired phenotype, and their corresponding genetic sequence is amplified. This process is repeated until the desired level of activity is obtained.
One key to a successful BOI evolution experiment is to optimize this cycle to ensure efficient exploration of the BOI fitness landscape. In most cases, mutations are deleterious to a BOI and result in a lower BOI fitness. However, rare mutations result in slight improvements in the desired BOI function that increases the protein's overall fitness. Directed evolution experiments that efficiently scan sequence space for fitness improvements are more likely to find BOls with optimized functions. Some key parameters that
influence the efficiency of this search include 1) a starting BOI with desirable features such as thermostability and at least a modicum of activity towards the desired biological function; 2) a high rate of mutagenesis to scan a large area of sequence space; and 3) a strong screening or selection method that can amplify BOI variants that display improved fitness.
1. Mutag Gene Library
Gene of Interest 2. Expression Directed Evolution Cycle BOI Library Improved Gene 4. Amplification . Selection
Figure 1.1. Directed Evolution Cycle. 1. A gene of interest is mutagenized to form a gene library. 2. This library is then expressed as either an RNA or protein, depending on the target function, to form a BOI library. 3. Selection isolates more active BOI variants. 4. The improved gene corresponding to the more active BOI is amplified, and the cycle is repeated until the target functional activity is achieved.
Numerous directed evolution schemes that satisfy these parameters have been devised to evolve BOIs in vitro, in bacteria, or in yeast.4- Novel approaches to generating libraries based on homologous recombination in addition to mutagenesis, and new methods of selecting positive library members based on fluorescent screening or antibiotic selection have enabled the generation of new or improved functions in a variety of simple organisms.-'0 Recent advances have made directed evolution of BOIs directly in the metazoan cellular environment possible, although these approaches remain limited in scope and efficiency." Below, I highlight how these approaches have been used specifically to create biomolecules that function in the complex metazoan environment. I first discuss directed evolution platforms in simple systems, and their limitations in creating biomolecules that reliably function in eukaryotes. I then discuss directed evolution in metazoa, and the technological hurdles that need to be overcome in order to make metazoa a more practical environment for directed evolution of biomolecules.
1.2. In vitro directed evolution
The simplest environment for running a directed evolution cycle is the test tube.'2 In vitro systems give the
researcher the greatest level of control over library generation and screening to ensure efficient navigation of the BOI fitness landscape. Advances in mutagenesis methods provide many options for creating diverse BOI libraries that can target specific regions of genes, or randomly mutate the enitire sequence." Screening approaches that link the BOI's genetic sequence to its expressed phenotype have been developed for a multitude of diverse functions.7 However, in vitro systems are limited by the lack of specific features that are unique to biological environments, including macromolecular crowding, and a robust chaperone network.4" These features often result in BOIs that fail to function properly when transferred to metazoan systems.
1.2.1. In vitro mutagenesis
In vitro mutagenesis of biomolecules remains the most widely used approach for library diversification,
even when the screen or selection will be performed in living organisms. Several error-prone polymerase chain reaction (epPCR) protocol with altered levels of Mg2+/Mn2+ ions or dNTPs, or mutagenic DNA
polymerase mixtures are capable of introducing a high level of mutations throughout the gene of interest (Figure 1.2a).'6,"7 epPCR is useful if the BOI already displays a modest level of activity with respect to the desired function. The incremental changes generated by random mutagenesis allows the BO to explore the fitness landscape in a stepwise manner, and can efficiently identify pathways to greater fitness.
If sequence information is available regarding how specific residues in a BOI influence function, targeted mutagenesis approaches can allow for more efficient scanning of a narrower sequence space that is more likely to affect the BOIs function. In site-saturation mutagenesis, mutagenic primers containing codons for every amino acid are used to mutate specific positions within a protein sequence.'8 Site-saturation mutagenesis will generate libraries that encompass the totality of sequence space for specific
positions. This approach allows researchers to specifically target positions that have a greater chance of improving fitness, while simultaneously ignoring residues that are either not important for the desired function, or are required for biophysical stability.
Other in vitro mutagenesis strategies use recombination to mix and match DNA sequences of different lengths. One strength of using recombination as a tool to generate diversity is that recombinatorial libraries allow beneficial mutations that may otherwise compete with one another on separate strands during screening/selection to combine, potentially resulting in a BOI with greater fitness than either single mutation alone." Recombination-based approaches can also combine library elements from different BOIs to generate combinations that can result in interesting new properties. 9 The most common in vitro recombinatorial diversification approach is DNA shuffling (Figure 1.2b).0 In DNA shuffling, a BOI gene is digested with DNase to generate short DNA fragments. These fragments are then randomly ligated together based on sequence homology and extended via PCR. Many recombination-based library
diversification strategies rely on enzymatic digestion of the DNA template followed by re-ligation.' Other approaches use modified PCR-based strategies to create partially amplified sequences that can then recombine to generate new sequence patterns.2 3
1.2.2. In vitro screening and selection for functional BOIs
Any successful screening or selection approach requires an effective mechanism to couple a BOI's genetic sequence to its phenotype.7 Once a phenotype displaying greater fitness is selected, the corresponding genetic sequence must be amplified in order to assess the sequence-level mutations that resulted in the improved phenotype. Cells provide a natural genotype/phenotype linkage in the form of intracellular transcription and translation machinery to express the BOI from its gene, and membranes to spatially separate the gene and BOI from other variants. In vitro screening and selection systems do not have this benefit, and require alternative methods of linking the genotype and phenotype of individual library members while still allowing large libraries to be screened efficiently.
a DNA Polymerase
b
Anneal Fragment aussnExtendua EM==
M=
""""" fDenature Amplify
full-length genes
by PCR
Figure 1.2 Strategies for in vitro mutagenesis. a) Error-prone PCR (epPCR) uses either a mutagenic polymerase mixture, uneven dNTP concentrations, and/or additional Mg2+
or Mn2
+ to introduce mutations (red) into a PCR amplicon. b) DNA shuffling begins with
fragmentation of genes of interest via DNAse I. Repeated cycles of annealing, extension, and denaturation will yield recombinatorial libraries based on short overlaps between fragments. The full-length gene is then amplified by PCR to generate recombined genes of interest (adapted from Packer et. al).7
In vitro screening and selection systems are also limited in terms of BOIs that can be evolved for
functions in metazoan cells. The in vitro environment, usually an aqueous buffer, lacks many of the complex cellular components that allow certain functions to be evolved. In vitro screening typically requires a fluorescent output so that spatially-separated variants can be screened via a plate reader.4
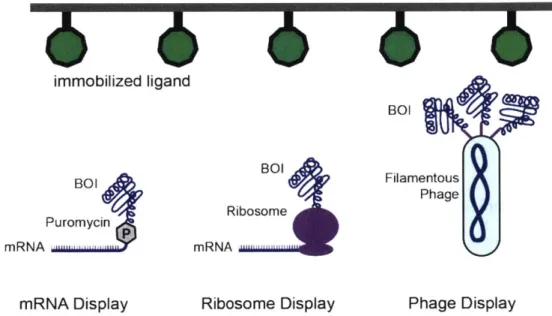
More recently, water-in-oil-in-water emulsions have enabled fluorescence-activated sorting of compartmentalized BOIs to to screen even larger library sizes.2 5 However, the constraint imposed by simple-to-screen metrics such as fluorescence confines most in vitro screens to BOIs such as metabolic enzymes and fluorescent proteins.26,27 While in vitro screening approaches are generally limited to metabolic enzymes and fluorescent proteins, in vitro selection approaches are limited to molecular binding interactions. These approaches covalently couple the BOI variant to its corresponding genetic sequence via either mRNA, the ribosome, or filamentous phage (Figure 1.3).2830 These approaches all covalently attach the protein of interest to the genetic sequence, and display the protein to bind to an immobilized ligand. Washing away low affinity binders leaves behind BOIs that bind the target ligand, and the corresponding linked genetic sequence can be amplified and sequenced. In mRNA display, a puromycin-containing linker is added after in vitro
transcription of a diverse library, but before in vitro translation. The puromycin covalently links the mRNA transcript to the resulting translating protein, facilitating amplification after selection. In ribosome display,
in vitro translation is halted after the protein is translated by the absence of a stop codon, and by increasing
the concentration of magnesium, resulting in both the mRNA transcript and folded protein of interest attached to the ribosome. Finally, in phage display, a mutational library is expressed on the exterior of a filamentous phage. After selection, the genes corresponding to successful binders are amplified from the phage genome.
immobilized ligand
BOI
BOI BOI Filamentous
Phage Ribosome
Puromycin
mRNA ... mRNA ,..
mRNA Display Ribosome Display Phage Display
Figure 1.3. In vitro selection by protein display. Three different display techniques are
used to select for binders to an immobilized target ligand. All three approaches, mRNA display, ribosome display, and phage display covalently link the BOI (in these cases, a protein) to its corresponding gene sequence. mRNA display and ribosome display are entirely in vitro techniques, while phage display requires expression and purification of phage from Escherichia coli (E. coli).
1.2.3. In vitro directed evolution of BOIs for function in metazoan cells
Several useful proteins have been evolved in vitro for use in metazoan systems. Most prominently, a combination of error-prone PCR and an in vitro screening platform have been used to evolve a number of useful fluorescent proteins across the color spectrum.7 Importantly, unlike the dimeric starting BOI, dsRed, many of these variants are monomeric. Monomeric fluorescent proteins are particularly useful for probing intracellular localization.' Phage display has been utilized extensively to evolve and optimize antibody
sequences for therapeutic production.'"' The best-selling biotherapeutic to date, adalimumab, was originally optimized for binding against immobilized TNFc via phage display of a human FAB (fragment antigen binding).2 Antibodies have also been optimized against the HIV-gp120 protein with picomolar
binding affinity, highlighting the ability of phage display to isolate high affinity binders."
The inherent limitations posed by the lack of a biological environment limits in vitro directed evolution systems to functions that can be assayed biochemically in a high-throughput manner. Furthermore, the unique biological environment of metazoan systems is difficult to replicate in vitro owing to factors such as complex proteostasis networks, macromolecular crowding, a multitude of post-translational processing enzymes, and subcellular compartmentalization. Consequently, BOls evolved in
vitro often fail to function when transferred to metazoan systems. It is therefore clear that there are many
advantages to evolving BOIs in vivo that simply cannot be resolved by in vitro directed evolution.
1.3. Directed evolution in bacteria and yeast
Compared to in vitro directed evolution, an entirely different class of biological functions can be evolved
in vivo. Biological organisms such as Escherichia coli (E. coli) or Saccharomyces cerevisiae (S. cerevisiae)
are already compartmentalized, which immortalizes the genotype/phenotype linkage and facilitates screening approaches such as fluorescence-activated cell sorting (FACS)." Furthermore biological organisms present a straightforward approach to the selection of BOI variants with greater fitness by coupling improvement of the BOI phenotype to survival of the organism. These approaches have greatly expanded the repertoire of biological functions that can be evolved to any BOI whose function can be coupled to expression of a necessary gene, or suppression of a toxic gene."
Despite these benefits, in vivo directed evolution presents its own problems. In vivo mutagenesis is more complicated owing to the low tolerance for mutations in biological organisms.3 5
Furthermore, biological organisms are constantly exploring organismal fitness landscapes that can "cheat" the engineered selection criteria and allow the organism to survive in spite of the engineered selection schemes. Finally,
biological organisms take time to grow and amplify positive BOI variants, which, especially under auxotrophic selection, can lengthen the amount of time required to evolve a desired phenotype. New strategies that enable in vivo continuous directed evolution have alleviated some of the burden posed by slow selection and amplification, although these strategies present their own challenges.6 Below, I discuss current approaches for directed evolution in bacteria and yeast, and highlight key examples of BOIs evolved in these systems for function in metazoan cells.
1.3.1. Directed evolution in bacteria for function in metazoan cells
The high transformation efficiency and relative simplicity for genetic engineering make bacteria the most widely used host for directed evolution.3 6 Bacterial mutagenesis is typically accomplished by one of two approaches: mutator strains that are deficient in mismatch repair and DNA polymerase exonuclease activity, or mutator plasmids that induce cytidine deamination and inhibit base excision repair."," Unfortunately, the mutation rates of mutator strains, while higher than the basal mutation rate of E coli, are still low
compared to in vitro mutagenesis approaches. Mutator plasmids confer a much higher mutation rate, but a noteworthy tradeoff is that these plasmids induce a significant level of cytotoxicity, likely due to bacterial sensitivity to mutations. More recently, targeted mutagenesis approaches allow for efficient bacterial library diversification with minimized levels of bacterial cytotoxicity. One approach uses a fuision between rat APOBEC I and T7 RNA polymerase to perform somatic hypermutation (SHM) in a targeted manner (Figure 1.4).38 Termed "MutaT7" the T7 RNA polymerase rAPOBECI fusion generates a high degree of mutations between the T7 promoter and terminator sites, with minimal leakage into off-target sites. Another targeted mutagenesis approach uses a fusion between a nickase Cas9 (nCas9) and an error-prone nick-translating DNA polymerase variants, termed EvolvR." Unlike rAPOBECI, which can only mutate cytidines, EvolvR can mutate any base within a 15-350 base pair window, defined by nCas9-targeting guide RNAs (gRNAs) depending on the DNA polymerase fusion.
Selection in bacteria is typically accomplished via fluorescent screening or antibiotic selection. Enhanced GFP (eGFP; one of the most frequently used fluorescent protein in metazoan systems), was
evolved using FACS to screen for GFP variants that exhibited greater intensity." Antibiotic selection can be used to evolve BOIs via a genetic couple, whereby BOIs exhibiting improved activity induce expression of a selection marker that confers resistance to an antibiotic. When bacteria are plated on agar containing the antibiotic, only cells expressing improved BOIs can induce the resistance marker survive. Antibiotic selection has been deployed to evolve a few different activities for use in metazoan systems, including transcription factors, recombinases, and targeted base editors.10'4 0 2
-T7 RNAP
rAPOBEC1
T7 Promoter T7 Terminator Array
Figure 1.4 Targeted, processive in vivo mutagenesis. T7-RNA polymerase (T7 RNAP) will only transcribe
DNA between T7 promoter and T7 terminator sequences. Processive, targeted mutagenesis of a gene of
interest is accomplished by flanking the gene of interest with T7 promoter and a T7 terminator array, and fusing an rAPOBEC 1 effector domain to T7 RNAP. T7 RNAP transcribes the gene of interest between the
T7 recognition elements, enabling SHM by rAPOBEC I along the entire DNA sequence.
Despite the utility of these approaches, they still rely on researcher manipulation at all stages of the directed evolution cycle, including generation of a diverse library by in vitro mutagenesis or recombination approaches, transformation into bacteria cells, and amplification and cloning after screening/selection of improved variants. These methods can be labor intensive and time-consuming, meaning individual rounds of directed evolution can take days to weeks to complete. In contrast, continuous directed evolution strategies integrate the entire directed evolution cycle into a single methodology that minimizes the need for researcher intervention, and dramatically increases the throughput of directed evolution." Perhaps the most generally successful in vivo continuous directed evolution strategy, termed phage-assisted continuous evolution (PACE) couples replication of a filamentous bacteriophage to the fitness of an evolving BOI (Figure 1.5).6 Unlike normal bacterial directed evolution, which can take at least a week to run a single round, PACE can run dozens of rounds of directed evolution in a single day owing to the fast replication time of bacteriophage. PACE has been used to evolve a variety of BOs, DNA binding proteins, proteases, ligand binders, RNA-guided nucleases, and tRNA/aminoacyl-tRNA synthetases (tRNA/aaRS).4448
While E. coli has been used as a host for directed evolution of BOIs for function in metazoan systems, the prospects remain somewhat limited. Bacterial environments differ greatly from metazoan environments, and thus BOIs that are optimized in bacterial systems often fail to function when transferred to metazoan systems.49 Differences such as an intensive proteostasis network, intracellular compartmentalization, redox environment, and glycosylation can all confound and derail directed evolution experiments. It is well-appreciated that the reverse is also true: Metazoan BOIs that are transferred to E.
coli often fail to express properly owing to the lack of similar chaperone networks that assists in proper
protein synthesis and folding in metazoan systems.' Furthermore, many of the unique signaling networks involved in development, and chromatin regulation machinery that make metazoan cells interesting to study cannot be reproduced in bacterial systems. While E. coli is a versatile host for directed evolution of BOIs that function in vitro and in E. coli, it is frequently not suitable for directed evolution of BOIs that function
in metazoan cells.
vi) influx of fresh
host E. coli
vii) Phage replicate to avoid dilution
QDmmmf
(iii) functional BOI induces gill expression (i) M13 phage encoding
evolving BOI ** *
(iv) only functional variants propagate
(ii) Mutagenesis
plasmid introduces BOI diversity
(v) Non-functional BOl cannot induce gill expression
Figure 1.5. Phage-assisted continuous evolution (PACE). i) M13 bacteriophage encoding the BOI infect E. coli. ii)
a mutagenesis plasmid introduces diversity into intracellular DNA, including the phage-encoded BOI. iii) selection is accomplished by coupling BOI activity to functional expression of gill. If the BOI can induce expression of gene Ill, infectious phage will be produced, iv) which can then go on to infect new cells. v) if the BOI cannot induce gIl expression, no gene III will be produced, and the phage will be non-infectious. vi) new host E. coli are continuous introduced into the culture to provide new hosts for replicating phage. vii) both cells and phage are removed as waste, facilitating selection of phage that can replicate fast enough, via gene III induction, to avoid dilution.
1.3.2. Directed evolution in yeast for function in metazoan cells
Directed evolution in yeast such as S. cerevisiae resolves many issues associated with developing BOIs for function in metazoan systems. Yeast cells have a nucleus and chromatin organized by nucleosomes. Yeast and metazoa both have membrane-bound organelles with similar structure and function. Furthermore, as there are robust systems for directed evolution, yeast is frequently employed to evolve BOIs for function in metazoan cells.
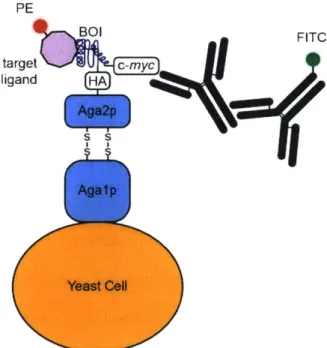
The most common approach for selection in yeast is cell surface display (Figure 1.6).' In yeast surface display, a BOI is fused to the C-terminus of the Aga2p subunit of yeast surface protein a-agglutinin. This protein fusion displays the BOI out away from the yeast cell surface where it can either interact with ligands, or be detected via a combination of specific antibodies and FACS. Yeast surface display has been used extensively for affinity maturation of single-chain variable fragments (scFvs), FABs, and for directed evolution of small molecule binding scaffolds." Yeast surface display is also used for directed evolution of catalytic enzymes, provided that the chemical product can be efficiently detected and coupled to corresponding the yeast cell.2 5
Yeast surface display has proven to be remarkably robust at screening for BOIs with improved functions. The approach is still limited by the time-consuming nature of in vitro library generation, and the poor transformation efficiency of S. cerevisiae with respect to E. co/i.' To generate libraries in vivo in yeast, researchers have begun developing mutagenesis systems based on self-replicating recombinatorial libraries, or orthogonal DNA replication.8,56 Recent reports suggest that orthogonal replication can generate mutations at a similar rate to state-of-the-art mutagenesis platforms, and could be used to evolve many new types of BOIs in yeast (unpublished results).57
While S. cerevisiae is now frequently used as a host organism for directed evolution of BOIs for function in metazoan cells, there are still some significant limitations. Yeast surface display is a useful approach for evolving certain types of activities, but evolution of many catalytic and intracellular processes remains elusive. While yeast N-hybrid systems have been extensively used to report on in vivo interactions,
these systems have not been generally co-opted for directed evolution.5 8 Furthermore, while S. cerevisiae has many important biological similarities with metazoan systems, there are still key differences related to protein trafficking, cell signaling, proteostasis, and post-translational modifications that can disrupt directed evolution experiments intended to evolve BOIs for function in metazoan systems.59 Finally, yeast-based directed evolution schemes still generally require in vitro genetic manipulation to design the mutational library, and clone enriched variants. These manipulations take substantial time and analysis to validate at each stage, which can limit experimental throughput and the researcher's ability to efficiently search sequence space. PE BOI FITC target C-myc ligand HA Ag a2p S
El
Agaip Yeast CellFigure 1.6. Yeast surface display. A protein of interest is fused to the Aga2p subunit of a-agglutinin, which is expressed on the yeast cell surface, facing away from the cell. Typically, functional variants are selected via FACS, based on simultaneous signals from two fluorophores: signifying both BOI expression on the yeast cell surface, and ligand binding. More recently yeast surface display has been used to screen for catalytic enzymes.
1.4. Directed evolution in metazoan systems
The various drawbacks of evolving BOIs for function in metazoan systems via in vitro, bacterial or yeast systems highlight the need for robust methods for evolving BOIs directly in the metazoan environment.
However, metazoan cells pose unique problems for running directed evolution cycles. Metazoan cells have a low mutation rate, making it difficult to generate or test libraries in a metazoan host. Additionally, their ability to take up and replicate multiple DNA sequences either by transfection or transduction makes it more difficult to link a desired phenotype with a specific genotype. Finally, their slow growth rate limits their ability to quickly and efficiently perform selections and amplify library members. Below, I highlight the most prominent approaches taken by researchers to address the problems posed by mutagenesis, selection, and amplification in complex eukaryotes.
1.4.1. Mutagenesis in metazoan systems
In vivo library diversification poses unique challenges for metazoan systems, owing to a low tolerance for
both chemical mutagens and the need to inhibit complex repair pathways. Global mutagenesis approaches generally cause increased cytotoxicity, and can result in cheating pathways, so researchers often seek targeted mutagenesis approaches. Fortunately, metazoan have developed their own approach to generate biomolecule libraries in an efficient and targeted manner as part of the adaptive immune system. After exposure of a B cell to an antigen, the variable regions of antibodies acquire mutations at a high rate via somatic hypermutation (SHM).60 Somatic hypermutation is mediated by cytidine deaminase (AID), which deaminates cytosine bases to uracil (Figure 1.7).61 If the uracil is not recognized by repair pathways, DNA synthesis results in C-T (or G4A) transitions which comprise >40% of all mutations generated by SHM.62
Uracil can also be recognized by Uracil-DNA glycosylase (UGI), which cleaves the Uracil base generating a DNA adduct. While this process can then result in insertion of cytidine by template-directed repair, DNA replication can also result in unbiased insertion of any of the four nucleotides. Taken together, the effects of AID activity result in an error rate of -10-' mutations per base per generation, 106-fold higher than the normal mutation rate of complex eukaryotes.6 3
The first evidence that SHM could be targeted to non-Ig sequences was provided by an increased rate of reversion of a premature stop codon in a neomycin resistance marker that was placed in the Ig variable region.64 It was later demonstrated that SHM is likely targeted to Ig variable regions by
transcription, indicating that it was perhaps possible to mutate non-Ig proteins by simply introducing them into B cells under a strong promoter.65,66 Researchers therefore began to leverage SHM in B cells to generate mutational libraries in transgenes for directed evolution in metazoan systems.
Molecular change Resulting DNA
NH2
o N
O 'N '
OH Cytidine
Cytidine Deaminase (AID)
0
o NO O T
-0- -0 A
OH Uracil
Uracil Glycosylase (UGI)
O OH
-o- -O OH
Apyrimidic base
Figure 1.7. Mutagenesis through somatic hypermutation. Cytidine deaminase converts cytidine to uracil, resulting in C->T transitions. If the uracil is recognized by uracil glycosylase, the uracil base is cleaved from the sugar resulting in an apyrimidic base. This change can result in random nucleotide
incorporation during repair.
The first directed evolution experiments completed in B cells combined SHM and fluorescence-activated cell sorting (FACS) to evolve fluorescent proteins. The 18-81 mouse pre-B cell line was used to mutate GFP that was inactivated either by a premature stop codon, or a disabled fluorophore.6 7 68 Interestingly, treatment with trichostatin A, a histone-deacetylase inhibitor, further increased SHM, highlighting the important affects that chromatin remodeling can have on directed evolution in metazoans.69
A human cancer cell line, Burkitt lymphoma Ramos B cell line, was used to evolve a red-shifted RFP protein.70 While most of these RFP variants had either G-*A or C--T mutations, T-*G and T--A mutations were also observed, indicating that the expected UGI-mediated base excision repair pathways were still active on non-Ig transgenes.
While initial directed evolution by SHM experiments were conducted in either mouse or human B cell lines, the long doubling time of these cell lines (24h) made extended directed evolution experiments inefficient. Researchers therefore transitioned to using DT40 chicken B cells deficient in XRCC2.7
' DT40 cells typically diversify their IgM antibodies by a template-directed mutagenesis mechanism termed gene conversion. However, ablation of XRCC2 was shown to convert mutagenesis to constitutive somatic hypermutation.72 DT40 cells have a number of advantages as a vehicle for SHM-based directed evolution: they have a high mutation rate, are simple to genetically engineer, and, most importantly, divide rapidly for metazoan cells (12 h). Thus, by using DT40 cells researchers can deploy SHM-based directed evolution in a more efficient manner.
Directed evolution in DT40 cells was further enabled through the development of a plasmid system for direct integration into the Ig variable region termed pHypermut2.3 pHypermut2 incorporates BOIs for directed evolution into the Ig locus by homologous recombination. This system was used to enhance the fluorescence intensity of eGFP > 3-fold relative to the wild-type protein. The DT40 cell line has also been used to evolve non-Ig, non-fluorescent protein using a combination of SHM and cell surface display. This approach has been used to evolve ligand binding antagonists, and to perform affinity maturation of scFvs.74,75 In the latter case, SHM alone was not sufficient to generate biomolecule variants exhibiting the desired functions. Instead, a combination of previously generated scFv libraries combined with SHM was able to expand the overall diversity of the library enough to isolate scFvs with greater binding affinity.
Recent advances in chemical biology have freed somatic hypermutation from B cells, enabling targeted mutagenesis in any cell line. The recent CRISPR revolution has engendered several new chemical biology tools for editing DNA and modulating gene expression inside the cel.76-78 The power of Cas9 systems lies in the ability of RNA to guide the Cas9 protein to specific regions of DNA through