Donor-Acceptor Iptycenes with Thermally
Activated Delayed Fluorescence
The MIT Faculty has made this article openly available.
Please share
how this access benefits you. Your story matters.
Citation
Voll, Constantin-Christian A. et al. “Donor-Acceptor Iptycenes with
Thermally Activated Delayed Fluorescence.” European Journal
of Organic Chemistry 2017, 32 (August 2017): 4846–4851 © 2017
WILEY#VCH Verlag GmbH & Co. KGaA, Weinheim
As Published
http://dx.doi.org/10.1002/ejoc.201700703
Publisher
Wiley Blackwell
Version
Author's final manuscript
Citable link
http://hdl.handle.net/1721.1/114245
Terms of Use
Creative Commons Attribution-Noncommercial-Share Alike
COMMUNICATION
Donor-Acceptor Iptycenes with Thermally Activated Delayed
Fluorescence
Constantin-Christian A. Voll,
[a]Jens U. Engelhart,
[a]Markus Einzinger,
[b]Marc A. Baldo
[b]and Timothy
M. Swager
*
[a]Abstract: A new donor-acceptor iptycene containing carbazole
donors and a thiadiazoloquinoxaline acceptor was synthesized and its photo- and electrochemical properties evaluated. The key intermediate 1 allows a lateral modification through cross-coupling and the (triisopropylsilyl)acetylene product 2 exhibits bright yellow fluorescence with emission lifetimes of 2.42 s in deoxygenated hexane. The long lifetime and high quantum efficiency (73%) is quenched by O2, and therefore attributed to thermally activated delayed fluorescence (TADF). This approach allows functionalization through cross-coupling reactions and depicts a promising scaffold for the synthesis of TADF-active molecules.
Introduction
There has been extensive research into OLED technologies from academia and industry in recent years, as a result of the advantages over traditional liquid crystal displays such as flexibility,[1] higher contrast ratio, and ease of manufacturing.[2]
However there is an inherent drawback with fluorescent organic materials in that singlet and triplet excitons are obtained in a 1:3 statistical ratio upon electrochemical excitation.[3] Since only the
radiative decay from the singlet state is spin-allowed, the internal electroluminescent quantum efficiency (int) is limited to 25%.
Heavy metal luminescent complexes containing iridium or platinum circumvent this issue through efficient spin-orbit coupling and have been shown to achieve int of virtually 100%.[4,5]
Phosphorescent organometallic materials have therefore played a vital role in the development of OLED technologies but suffer from drawbacks including cost, element sustainability, stability of blue emitters, and triplet-triplet annihilation at strong current densities causing quantum efficiency roll-off.[6] Delayed
fluorescence and emission from the doublet state by stable emissive radicals have been proposed as two triplet harvesting alternatives.[7,8] Delayed fluorescence can also in principle, be
achieved through the up-conversion of triplet excitons to singlet excitons by triplet-triplet annihilation (TTA),[9] hybridized local
charge transfer (HLCT)[10] or thermally activated delayed
fluorescence (TADF).[11–13] TADF tends to outperform the other delayed strategies and has recently been reported to achieve high
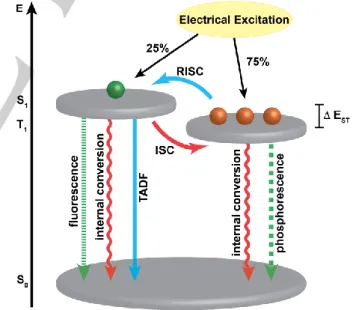
emission efficiencies, thus rendering this approach a promising strategy to create efficient OLEDs.[14–16] In TADF materials, intersystem crossing is very fast allowing for the thermal equilibration of triplet and singlet excitons (Figure 1). This can be realized when the S1 and T1 states are close in energy because
of the inverse proportionality between the singlet-triplet gap (EST)
and the first-order mixing coefficients.[17] Adachi and co-workers
have pioneered TADF and have demonstrated that introducing a torsional angle between donor and acceptor groups results in largely segregated HOMO and LUMO states that lower the exchange energy and give rise to a small EST.[15,18–23] Our group
recently reported an alternative approach with which TADF can be achieved through homoconjugation in donor-acceptor tripcycenes.[24] We initially chose the triptycene scaffold as a
result of its inherent high thermal stability,[25,26] a vital factor in the
OLED manufacturing process. We seek to expand this approach to materials that allow for a more diverse functionalization to modulate emission wavelength and realize higher quantum efficiencies.
Figure 1. Competing photophysical processes from the excited state. isc =
intersystem crossing, risc = reverse intersystem crossing. Excitons visualized as balls. Drawing inspired by Adachi and co-workers, Ref. 17.
The design of new iptycene donor-acceptor structures utilizes known building blocks. We anticipated that carbazole, a strong donor that can be substituted with solubilizing alkyl side chains, can be paired with a thiadiazoloquinoxaline acceptor to produce a TADF material (Figure 2).
[a] Department of Chemistry
Massachusetts Institute of Technology
77 Massachusetts Avenue, Cambridge MA, 02139, USA E-mail: [email protected]
[b] Department of Electrical Engineering and Computer Science Massachusetts Institute of Technology
77 Massachusetts Avenue, Cambridge MA, 02139, USA
revised.docx
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
COMMUNICATION
Figure 2. Structure of the donor-acceptor iptycene along design principles.
As shown in Figure 3, the HOMO and LUMO frontier orbitals are spatially well separated promoting a small EST and enhanced T1 S1 reverse intersystem crossing. The HOMO is delocalized
over the carbazole moieties whereas the LUMO is located on the thiadiazoloquinoxaline acceptor suggesting that the S1 state has
charge transfer character. Whilst heavy atoms are not required for an efficient spin conversion, we anticipated that the introduction of the sulfur atom in the thiadiazole moiety may still be favorable for higher intersystem crossing rates. Such processes are expected to enhance the interconversion of T1 and S1, and that
the rigid framework minimizes non-radiative pathways from the excited states.
Figure 3. Calculated HOMO and LUMO of 2 without alkyl chains based on
B3LYP functional and 6–31+G* basis set.
Results and Discussion
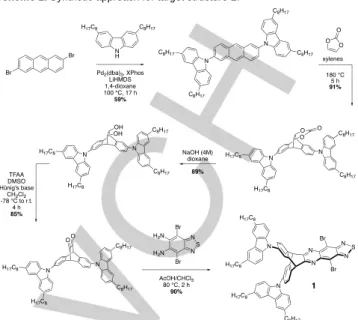
Our target core molecule, 1, was synthesized as shown in Scheme 1, starting with a Pd-catalyzed N-arylation of 3,6-dioctyl-9H-carbazole with 2,6-dibromoanthracene using XPhos as the ligand. Other conditions including different palladium sources, ligands or Ullmann coupling failed to do this transformation in reasonable yields. With the dicarbazoleanthracene intermediate in hand, the diketone structure was obtained by a Diels-Alder cycloaddition with vinylene carbonate, followed by a basic hydrolysis and a Swern oxidation, all proceeding in high yields. The final step consisted of the condensation with 4,7-dibromobenzo[c][1,2,5]thiadiazole-5,6-diamine to form 1 in 90% yield.
Scheme 1. Synthetic approach for target structure 1.
Having prepared 1 with two opposing aryl bromides allows numerous modifications through cross-couplings. We chose a Sonogashira coupling with (triisopropylsilyl)acetylene to yield 2 in 76% yield (Scheme 2). Compounds 1 and 2 were characterized by 1H and 13C NMR, and high resolution mass spectrometry. Scheme 2. Final synthetic step toward 2.
With product 2 in hand, we were interested to evaluate its photophysical properties. We observed an absorption maximum at 478 nm with a pronounced shoulder peak at 508 nm in hexane (Figure 4), suggesting the expected charge transfer character. The compound is emissive with a maximum luminescence at 524 nm and a shoulder peak at 552 nm, manifested in a bright yellow emission. The preservation of the shoulder in the emission spectrum indicates that there is little reorganization in the excited state, presumably a result of the rigid framework. The Stokes shift of the S1 state is relatively small compared to typical charge
transfer states, indicating that the excitation involves a limited relaxation of the molecular geometry. We also noticed that the photoluminescence is sensitive to the polarizability of the solvent. In toluene, the emission is red-shifted by 76 nm (λem = 600 nm),
whilst in ethanol the emission peak is located at 548 nm. These more polarizable solvents negatively affect the quantum yields, as also seen in other donor-acceptor iptycenes (Figure S11).[24]
HOMO
LUMO
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
Figure 4. Photophysical characterization of 2 in hexane. Red curve shows molar
absorptivity and orange curve the emission spectrum.
To evaluate if this iptycene scaffold is TADF active, we sought out the effects of oxygen that quenches the long-lived triplet states and were looking for both a nanosecond scale prompt emission as well as a microsecond scale delayed component. The compound showed a high quantum yield of 73% in deoxygenated hexane, much higher than the previously reported homoconjugated triptycenes from our group,[24] that
decreased to 43% after oxygen saturation. This observation suggests that there is a long-lived excited state that is quenched by oxygen indicative of TADF behavior. Time-resolved photoluminescence with a streak camera was employed (Figure 5) and the results show the expected microsecond response (2.42
s lifetime), which disappears to a nanosecond emission lifetime after O2 saturation (12.5 ns lifetime). Both findings are evidence
that this molecule is indeed TADF active. It is furthermore noteworthy that higher concentrations quench the emission and that the relative peak heights change in favor of a longer wavelength emission at higher concentrations, indicating excimer formation (Figure S13). The absorption spectra are independent of concentration and therefore don’t indicate the presence of aggregated species in the ground state (Figure S12).
Figure 5. Photoluminescence transition lifetime measurements in hexane at
room temperature.
The derivative of 2 without carbazole moieties has recently been published by Bunz and co-workers, and exhibits a similar
absorption and emission profile with a weaker charge-transfer character and slightly lower quantum yield.[27]
The HOMO energy levels were estimated using cyclic voltammetry (Figure 6). Compound 2 displayed a reversible oxidation wave at 0.76 V (vs Fc/Fc+), which can be assigned to
an oxidation of the carbazole moieties. The HOMO/LUMO levels were based on the oxidation potential and the onset of UV-vis absorption spectra and calculated as – 5.9/–2.0 eV.
Figure 6. Cyclic voltammogram of 2 in CH2Cl2 containing 0.1 M TBAPF6 as the
electrolyte. Scan rate: 100 mVs-1.
Conclusions
We have designed a high-yielding synthetic approach to the versatile intermediate 1 possessing a thiadiazoloquinoxaline acceptor, electron-rich iptycene wings and solubilizing alkyl chains. This privileged intermediate allows further functionalization using different cross-coupling partners due to two lateral aryl bromides. The (triisopropylsilyl)acetylene product
2 exhibits bright yellow fluorescence with emission lifetimes of
2.42 s and a quantum yield of 73% in degassed hexane. The long lifetime is quenched by O2 and is attributed to thermally
activated delayed fluorescence (TADF).
Experimental Section
All reagents and solvents were purchased from commercial sources and used as received. 1H and 13C NMR spectra were recorded in CDCl3 or
CD2Cl2 solutions on a Bruker AVANCE 400 MHz or JEOL ECZ-500
spectrometer. The chemical shift data are reported in units of δ (ppm) relative to residual solvent. Ultraviolet-visible absorption spectra were measured with a Cary 4000 UV-Vis spectrophotometer from Agilent Technologies and corrected for background signal with a solvent filled cuvette. All electrochemical measurements were carried out with Autolab PGSTAT30 potentiostat (Eco Chemie B.V.) in a conventional three- electrode configuration system: a platinum working electrode (1.6 mm diameter), a platinum wire counter electrode and a silver wire in 0.01 mM silver nitrate in acetonitrile with 0.1 mM nBu4PF6 as electrolyte in a porous
vycor fritted electrode as pseudo-reference electrode with ferrocene added after every run as the internal standard. Dichloromethane was employed as the solvent with 0.1 M nBu4NPF6 (TBAPF6) as the electrolyte and the
experiments were performed under ambient condition with a scan rate of
-250 -200 -150 -100 -50 0 50 100 150 200 -2.5 -1.5 -0.5 0.5 1.5
Cur
ren
t
(µA)
Potential vs Fc (V)
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
COMMUNICATION
100 mV/s. All DFT computations were carried out using the Spartan ‘10 software package. The gas-phase ground state geometry optimizations were performed using the B3LYP exchange-correlation functional in the 6-31+G* basis set.
3,6-dioctyl-9H-carbazole was synthesized according to a literature procedure.[28]
2,6-bis(3,6-dioctyl-9H-carbazol-9-yl)anthracene: In a heat gun dried
Schlenk tube under an atmosphere of argon, a mixture of dibromoanthracene (1.30 g, 3.87 mmol), dioctylcarbazole (2.86 g, 7.30 mmol,1.9 equiv.), Pd2(dba)3 (239 mg, 261 µmol, 7 mol%) and XPhos (278
mg, 583 µmol, 15 mol%) was dissolved in dry 1,4-dioxane (0.1 M, 39 mL). The resulting mixture was degassed in a stream of argon for 10 minutes. At this point LiHMDS (0.1 M in THF, 9.7 mL, 9.70 mmol, 2.5 equiv) was added. After heating at 80 °C for 17 h the reaction mixture was dissolved in CH2Cl2 and subsequently washed with water and brine. After drying over
MgSO4 and evaporating under reduced pressure, flash column
chromatography (silica gel, hexane/CH2Cl2 9:1) gave the product as a
bright yellow solid (59%, 2.20 g, 2.30 mmol). 1H NMR (400.05 MHz, CD 2Cl2, 25 °C): δ = 8.60 (s, 2H), 8.25-8.23 (m, 4H), 7.98 (d, 4H, J = 1.7 Hz), 7.73 (dd, 2H, J = 9.1 Hz, J = 2.0 Hz), 7.49 (d, 4H, J = 8.4 Hz), 7.27 (dd, 4H, J = 8.4 Hz, J = 1.7 Hz), 2.82 (t, 8H, J = 7.6 Hz), 1.70-1.79 (m, 8H), 1.46-1.28 (m, 40H), 0.90 (t, 8H, J = 6.9 Hz); 13C {1H} NMR (100.60 MHz, CD 2Cl2, 25 °C): δ =139.9, 135.8, 135.3, 132.5, 131.3, 130.4, 128.5, 127.6, 127.1, 126.7, 125.9, 124.8, 124.0, 120.0, 110.0, 36.4, 32.8, 32.4, 30.0, 29.8, 29.8, 23.1. 2,6-bis(3,6-dioctyl-9H-carbazol-9-yl)-9,10-[4,5]epidioxoloanthracen-13-one: In an oven dried pressure vessel dicarbazol anthracene (2.10 g,
2.19 mmol) was dissolved in xylenes (6 mL) and the resulting solution was degassed in a stream of argon for 10 minutes. An excess of vinylene carbonate (5 mL, 6.80 g, 79.1 mmol, 36 equiv) and a catalytic amount of hydroquinone (spatula tip) were added. The reaction was stirred for 24 hours at 180°C in the dark. At this point the yellow colored solution with blue fluorescence turned into a light brownish non-fluorescent solution. After evaporation of the reaction mixture under reduced pressure, the product was obtained after column chromatography (SiO2, hexane:CH2Cl2
1:1) as a colorless solid (2.09 g, 2.00 mmol, 91%). 1H NMR (400.05 MHz,
CD2Cl2, 25 °C): δ = 7.94 (d, 2H, J = 3.2 Hz), 7.70-7.63 (m, 4H), 7.54-7.48 (m, 2H), 7.35-7.31 (m, 2H), 7.29-7.20 (m, 6H), 5.08 (s, 2H), 4.91 (s, 2H), 2.84-2.76 (m, 8H), 1.77-1.67 (m, 8H), 1.44-1.28 (m, 40H), 0.90 (t, 8H, J = 6.8 Hz); 13C {1H} NMR (100.60 MHz, CD 2Cl2, 25 °C): δ = 154.2, 140.0, 139.7, 138.8, 138.4, 138.0, 136.3, 135.6, 135.4, 135.1, 128.6, 127.7, 127.1, 127.1, 126.6, 126.5, 125.6, 124.4, 123.9, 123.8, 120.0, 119.9, 109.7, 109.7, 76.6, 76.5, 48.0, 48.0, 36.3, 36.3, 32.8, 32.4, 30.0, 29.8, 29.8, 23.1, 14.8. 2,6-bis(3,6-dioctyl-9H-carbazol-9-yl)-9,10-ethanoanthracene-11,12-diol: The dioxolanone (2.00 g, 1.92 mmol) was dissolved in 1,4-dioxane
(20 mL) and a 4M aqueous solution of sodium hydroxide (7 mL) was added. The resulting mixture was refluxed for 1.5 h. After cooling to ambient temperature the product was extracted using chloroform (3 x 50 mL). Subsequently the combined organic layers were washed with aqueous NH4Cl (100 mL) and brine (50 mL). The crude product was obtained after
drying over magnesium sulfate and evaporation of the solvent. Column chromatography (silica gel, CH2Cl2) yielded the product as an off-white
sticky solid (1.74 g, 1.71 mmol, 89%). 1H NMR (400.05 MHz, CDCl 3, 25 °C): δ = 7.94 (s, 4H), 7.65 (d, 1H, J = 2.2 Hz), 7.60 (d, 1H, J = 7.8 Hz), 7.57 (d, 1H, J = 2.2 Hz), 7.54 (d, 1H, J = 7.8 Hz), 7.47 (dd, 1H, J = 7.8 Hz, J = 2.2 Hz), 4.60-4.56 (m, 2H), 4.26 (s, 2H), 2.81 (t, 8H, J = 6.7 Hz), 2.45-2.36 (brs, 2H), 1.79-1.70 (m, 8H), 1.44-1.29 (m, 40H), 0.91 (t, 12H, J = 6.8 Hz); 13C {1H} NMR (100.60 MHz, CD 2Cl2, 25 °C): δ = 140.1, 139.9, 138.9, 138.5, 138.3, 136.4, 135.8, 135.5, 135.3, 128.7, 128.7, 127.8, 127.2, 127.2, 126.8, 126.7, 125.8, 124.5, 124.1, 124.0, 120.1, 120.0, 109.9, 109.8, 76.8, 76.7, 48.2, 48.2, 36.5, 36.5, 32.9, 32.5, 30.2, 30.0, 29.9, 23.3. 2,6-bis(3,6-dioctyl-9H-carbazol-9-yl)-9,10-ethanoanthracene-11,12-dione: Trifluoroacetic anhydride (576 µL, 870 mg, 4.14 mmol) was added
at -78 °C to a mixture of dry DMSO (329 µL, 362 mg, 4.63 mmol) and dry CH2Cl2 (22 mL) under argon and the resulting solution was stirred for 10
min. A solution of the diole starting material (1.37 g, 1.35 mmol) in CH2Cl2/DMSO (2:1 (v:v), 10.5 mL) was added within 15 min and the
mixture was stirred for a further 60 min at -78°C. Hünig’s base (1.4 mL, 38.7 mmol) was added slowly, the reaction mixture was allowed to warm to room temperature and subsequently poured into 2M hydrochloric acid (200 mL). When the evolution of gas had stopped, the two layers were separated. The aqueous layer was extracted with CH2Cl2. The combined
organic layers were washed with water (50 mL) and dried over magnesium sulfate. The solvent was evaporated and the residue purified by flash column chromatography (silica gel, hexane:CH2Cl2 1:1) to give the
diketone as an orange solid (1.16 g, 1.18 µmol, 85%). The product was used in the next step without characterization due to its instability in solution.
4,13-dibromo-8,17-bis(3,6-dioctyl-9H-carbazol-9-yl)-6,11-[1,2]benzenobenzo[b][1,2,5]thiadiazolo[3,4-i]phenazine (1): Iptycene
quinone (975 mg, 962 µmol) and diamino benzothiadiazole (405 mg, 1.25 mmol) were dissolved in acetic acid (10 mL) and chloroform (7 mL). Upon heating to 80 °C the color changed to dark red as a consequence of product formation. After cooling to ambient temperature the reaction mixture was diluted with water and the product extracted with dichloromethane. The combined organic layers were washed with an aqueous solution of sodium carbonate and brine. After drying over MgSO4
and evaporating under reduced pressure, flash column chromatography (silica gel, hexane/ CH2Cl2 gradient of 3:1 to 1:1) gave the product as an
amorphous red solid (1.12 g, 90%). 1H NMR (400.05 MHz, CDCl 3, 25 °C): δ = 7.94 (d, 2H, J = 2.0 Hz), 7.89 (d, 2H, J = 7.8 Hz), 7.82 (d, 4H, J = 1.0 Hz), 7.47 (dd, 2H, J = 7.8 Hz, J = 2.0 Hz), 7.21 (d, 4H, J = 8.6 Hz), 7.09 (dd, 4H, J = 8.6 Hz, J = 1.5 Hz), 6.00 (s, 2H), 2.76 (t, 8H, J = 7.7 Hz), 1.73-1.64 (m, 8H), 1.41-1.24 (m, 40H), 0.91-0.84 (m, 12H); 13C {1H} NMR (100.60 MHz, CDCl3, 25 °C): δ = 158.6, 152.3, 142.3, 139.5, 138.7, 138.1, 135.1, 127.2, 126.8, 126.3, 124.2, 123.8, 119.8, 114.1, 109.5, 54.5, 36.2, 32.5, 32.2,29.8, 29.6, 22.9, 14.4. HRMS (ESI) m/z: [M+H]+ Calculated for
C78H88Br2N6S 1301.5235; Found: 1301.5253.
8,17-bis(3,6-dioctyl-9H-carbazol-9-yl)-4,13-
bis((triisopropylsilyl)ethynyl)-6,11-[1,2]benzenobenzo[b][1,2,5]thiadiazolo[3,4-i]phenazine (2): 1 (50 mg,
38.4 μmol), Pd(PPh3)4 (2.2 mg, 1.92 μmol) and CuI (0.4 mg, 1.92 μmol)
were suspended in HNiPr
2/THF (32 μL/ 0.3 mL) and degassed with argon
for 15 minutes before triisopropylsilylacetylene (37 mg, 88 μL, 380 μmol) was added and the resulting reaction mixture stirred at 45°C for 16 h. Upon completion, the reaction mixture was quenched with water and the product extracted with CH2Cl2, dried over MgSO4 and the solvent evaporated
under reduced pressure. The crude product was purified by column chromatography (SiO2, hexane/CH2Cl2 1:1) to give product 2 as a red solid
(43 mg, 76%). 1H NMR (500 MHz, Methylene Chloride-d 2) δ 7.91 (s, 4H), 7.85 (d, J = 7.8 Hz, 4H), 7.48 (d, J = 7.9 Hz, 2H), 7.30 (d, J = 8.3 Hz, 4H), 7.20 (dd, J = 8.4, 1.7 Hz, 4H), 5.79 (s, 2H), 2.77 (t, J = 7.7 Hz, 8H), 1.70 (p, J = 7.3 Hz, 8H), 1.29 (s, 90H), 0.87 (t, J = 6.7 Hz, 13H).13C NMR (126 MHz, CD2Cl2) δ 158.00, 154.53, 142.99, 141.58, 139.75, 139.54, 137.77, 135.23, 127.21, 126.99, 126.12, 124.04, 123.79, 119.86, 114.35, 109.72, 109.36, 101.33, 36.29, 32.73, 32.31, 30.08, 29.94, 29.75, 29.71, 18.99, 14.28, 11.93. HRMS (ESI) m/z: [M+H]+ Calculated for C
100H130N6SSi2 1503.9689; Found: 1503.9692.
Acknowledgements
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
Financial support was provided through the Airforce Office of Scientific Research, and the German Research Foundation (DFG) of JUE.
Keywords: TADF • donor-acceptor • iptycene • Sonogashira •
organic materials
[1] S. Kim, H.-J. Kwon, S. Lee, H. Shim, Y. Chun, W. Choi, J. Kwack, D. Han, M. Song, S. Kim, S. Mohammadi, I. Kee, S. Y. Lee, Adv.
Mater. 2011, 23, 3511–3516.
[2] C. A. Bower, E. Menard, S. Bonafede, J. W. Hamer, R. S. Cok,
IEEE Trans. Components, Packag. Manuf. Technol. 2011, 1, 1916– 1922.
[3] M. Segal, M. A. Baldo, R. J. Holmes, S. R. Forrest, Z. G. Soos,
Phys. Rev. B 2003, 68, 75211.
[4] C. Adachi, M. A. Baldo, M. E. Thompson, S. R. Forrest, J. Appl.
Phys. 2001, 90, 5048–5051.
[5] M. A. Baldo, D. O’Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson, S. R. Forrest, Nature 1998, 395, 151–154. [6] M. A. Baldo, C. Adachi, S. R. Forrest, Phys. Rev. B 2000, 62,
10967–10977.
[7] Q. Peng, A. Obolda, M. Zhang, F. Li, Angew. Chem. Int. Ed. 2015,
54, 7091–7095.
[8] A. Obolda, X. Ai, M. Zhang, F. Li, ACS Appl. Mater. Interfaces 2016,
8, 35472–35478.
[9] Y. Luo, H. Aziz, Adv. Funct. Mater. 2010, 20, 1285–1293. [10] W. Li, Y. Pan, R. Xiao, Q. Peng, S. Zhang, D. Ma, F. Li, F. Shen, Y.
Wang, B. Yang, et al., Adv. Funct. Mater. 2014, 24, 1609–1614. [11] K. Goushi, K. Yoshida, K. Sato, C. Adachi, Nat. Photonics 2012, 6,
253–258.
[12] H. Nakanotani, T. Higuchi, T. Furukawa, K. Masui, K. Morimoto, M. Numata, H. Tanaka, Y. Sagara, T. Yasuda, C. Adachi, Nat.
Commun. 2014, 5, 4016.
[13] L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang, Y. Ma, Angew.
Chem. Int. Ed. 2014, 53, 2119–2123.
[14] D. Y. Kondakov, T. D. Pawlik, T. K. Hatwar, J. P. Spindler, J. Appl.
Phys. 2009, 106, 124510.
[15] H. Uoyama, K. Goushi, K. Shizu, H. Nomura, C. Adachi, Nature
2012, 492, 234–238.
[16] L.-S. Cui, H. Nomura, Y. Geng, J. U. Kim, H. Nakanotani, C. Adachi,
Angew. Chem. Int. Ed. 2017, 56, 1571–1575.
[17] N. J. Turro, V. Ramamurthy, J. C. Scaiano, Modern Molecular
Photochemistry of Organic Molecules, University Science Books,
2010.
[18] A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki, C. Adachi, Appl. Phys. Lett. 2011, 98, 083302.
[19] G. Méhes, H. Nomura, Q. Zhang, T. Nakagawa, C. Adachi, Angew.
Chem. Int. Ed. 2012, 51, 11311–11315.
[20] H. Tanaka, K. Shizu, H. Miyazaki, C. Adachi, Chem. Commun.
2012, 48, 11392–11394.
[21] Q. Zhang, H. Kuwabara, W. J. Potscavage, S. Huang, Y. Hatae, T. Shibata, C. Adachi, J. Am. Chem. Soc. 2014, 136, 18070–18081. [22] S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N.
Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, et al., Nat.
Mater. 2015, 14, 330–6.
[23] Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki, C. Adachi, J. Am. Chem. Soc. 2012, 134, 14706–14709. [24] K. Kawasumi, T. Wu, T. Zhu, H. S. Chae, T. Van Voorhis, M. A.
Baldo, T. M. Swager, J. Am. Chem. Soc. 2015, 137, 11908–11911. [25] T. M. Swager, Acc. Chem. Res. 2008, 41, 1181–1189.
[26] H.-H. Chou, H. Shih, C. Cheng, J. Mater. Chem. 2010, 20, 798. [27] M. Ganschow, S. Koser, S. Hahn, F. Rominger, J. Freudenberg, U.
H. F. Bunz, Chem. Eur. J. 2017, 23, 4415–4421.
[28] X. Y. Wang, D. C. Yang, F. D. Zhuang, J. J. Liu, J. Y. Wang, J. Pei,
Chem. Eur. J. 2015, 21, 8867–8873.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
COMMUNICATION
Entry for the Table of Contents (Please choose one layout)
Layout 1:
COMMUNICATION
A new donor-acceptor iptycene scaffold is reported, with the key intermediate product allowing facile lateral modification through cross-coupling. The(triisopropylsilyl)acetylene product is highly emissive in solution and we demonstrate that this material is TADF active.
TADF, OLEDs
Constantin-Christian A. Voll, Jens U. Engelhart, Markus Einzinger, Marc A. Baldo and Timothy M. Swager* Page No. – Page No.
Donor-Acceptor Iptycenes with Thermally Activated Delayed Fluorescence