Delivery of Antigens to Dendritic Cells as a
Platform for a Vector-free Cancer Therapy
by Shirley Mao
SUBMITED TO THE DEPARTMENT OF MECHANICAL ENGINEERING IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
BACHELOR OF SCIENCE AT THE
MASSACHUSETTS INSTITUTE OF TECHNOLOGY JUNE 2014
Signature of
0 2014 Shirley Mao All Rights Reserved
k s-'
7/
greby
tiratto W pwm!esion to raproduce and toSignature redacted dt
publolypaper
and electronic copies of this thasi def&pv Au b erin prtW In any medumt now known or hereeftw crestd.Department of Mechanical Engineering May 23, 2014
Signature redacted
/
Accepted by
Klavs F. Jensen Warren K. Lewis Professor of Chemical Engineering Professor of Materials Science and Engineering
Signature redacted
Thesis SupervisorAnnette Hosoi Associate Professor of Mechanical Engineering Undergraduate Officer MASSACHUSETTS INSTITUTE OF TECHNOLOGY
JUL 3
0
2014
LIBRARIES
Certified byA5
CA
Delivery of Antigens to Dendritic Cells as a Platform
for a Vector-free Cancer Therapy
by Shirley Mao
Submitted to the Department of Mechanical Engineering on May 23, 2014 in Partial Fulfillment of the Requirements for the Degree of Bachelor of Science in
Mechanical Engineering
I.
Abstract
Cell based vaccines that activate a patient's immune system via an antigen-specific CD8 T cell response hold much therapeutic potential. One of the greatest challenges in the development of these vaccines is creating antigen presentation by delivery of antigens into the cell cytoplasm. It has been previously shown that a microfluidic chip developed at MIT is capable of intracellular delivery of macromolecules. We conducted a preliminary evaluation to determine whether an improved antigen-specific CD8 response can be achieved using the microfluidic squeezing platform.
Using this approach, we delivered proteins and antigens to bone marrow derived dendritic cells (BMDCs) and splenic dendritic cells. We initially delivered fluorescent dyes to confirm that intracellular delivery could be achieved. Then ovalbumin (OVA) was delivered as a model protein to assess the system's capability to prime dendritic cells against a particular antigen. Dendritic cells activated post-delivery were then cultured with isolated primary T cells in-vitro to determine whether a T cell response could be induced by the treated dendritic cells. The efficacy of the microfluidic treatment was assessed by measuring T cell proliferation activated by T cell receptor (TCR) binding to MHC Class I receptors presented on the treated dendritic cells. After verification of the ovalbumin model, we propose to move to use B16F1O melanoma cell lysate as an antigenic source, a more clinically representative antigen source.
Thesis Supervisor: Klavs F. Jensen
Title: Warren K. Lewis Professor of Chemical Engineering, Professor of Materials Science and Engineering
11.
Acknowledgements
Professors
Professor Klavs F. Jensen: I appreciate the continued interest he has shown in this project and the support he has shown his UROPs. I would also like to thank him for being extremely
supportive of mentoring and encouraging undergraduate research.
Professor Robert Langer: Thank you for your continued support for this project and for serving as an inspiration for many.
Mentors
Armbn Sharei: For his unwavering, continued support during the past three and a half years. He has exposed me to multiple facets of research and life and has single-handedly taught me more than any class could have. Thank you for your patience, enthusiasm, and knowledge. In you I have been lucky to find not only a great mentor but also a great friend.
Squishyurops
Nahyun Cho: For taking me under your wing and teaching me a lot about research and life. Thank you for always having my back.
Roberta Poceviciute: For always keepin' it real and for being perpetually awesome.
Emily Jackson: I miss you, and all the girly shopping times we had!
George C. Hartoularos: For always being positive, smiling, taking initiative and being enthusiastic, and the best little research brother.
Tanya Talkar: For being the best 'little' in research and life that I could ask for. Thank you especially for working with me on our challenging immunology project and sharing all the little victories.
Megan Heimann: Thank you for being the fearless leader of the immunology project.
The rest of the family: When Armon had 3 UROPs, we bought him a peapod. When he had around five, we said he was a momma duck. When he got around 8, it was an octopus. But now there are too many and you all split faster than my cells. Thanks for being so fun, and I'm going to miss you all like crazy. Keep squeezing!
Family
Suo-ya Chen, Weiwei Mao, Solon Mao: And thank you of course, to my awesome family for their continual love and support through my tough times at MIT and beyond.
Table of Contents
1. Introduction ... 8
1.1 Potential for Cancer Vaccines... 8
1.2 Current Methods of Antigen Delivery and Their Shortcomings ... 10
1.3 Rationale for New Intracellular Delivery Platform ... 11
2. CellSqueeze Platform Development ... 12
2.1 System and Device Design ... 12
2.2 U sing the System ... 14
2.3 Efficacy of the Device on HeLa Cells ... 15
2.3.1 Facs Data Analysis... 15
2.3.2 Defining Delivery Efficiency... 15
2.4 Assessment of Potential for Protein Delivery ... 16
2.5 SE M Studies ... 17
3. Using the CellSqueeze Platform to Enhance Antigen Presentation for Cancer Vaccination Development... 19
3.1 Assessment of the Delivery Potential of the CellSqueeze Platform to Primary Immune C ells ... 19
3.1.1 Delivery of Dyes to Primary Murine derived Dendritic Cells (Spleen) ... 19
3.2 Inducing Antigen Presentation In-vitro ... 20
3.2.1 OVA as a Model Antigen ... 20
3.2.2 OVA Delivery to Primary Murine Derived Dendritic Cells (Spleen) ... 20
3.2.3 OVA Delivery to Bone Marrow Derived Dendritic Cells (BMDCs) ... 21
3.3 Assessment of Normal Cell Function after Treatment by the Device ... 23
3.3.1 DC Maturation Studies ... 24
3.3.2 Effect of Device Treatment on MHC I and MHC II Pathways ... 25
4. In-vitro Assays for activating an Immune Response for a known Antigen...26
4 .1 C o-culture M odel ... 27
4.2 FACS: Measuring T cell Activation ... 27
4.3.1 Rationale & Advantages ... 29
4.3.2 B 16 OVA M elanom a Lysate M odel... 30
4.3.3 Lysate Dosage Testing for System Sensitivity ... 30
4.3.4 Lysate Toxicity Hypothesis ... 31
4.3.5 Lysate Toxicity A ssay... 32
4.3.6 Lysate Characterization ... 32
5. In-vivo Assays for Activating an Immune Response for a Known Antigen ... 32
5.1 In-vivo Proliferation Translation Assay... 32
6. Future work & Project O utlook ... 34
7. Appendix & M ethods...34
7.1 Preparation of SEM Sam ples ... 34
7.2 Running the Chip ... 35
7.3 Bone M arrow Dendritic Cell Creation... 35
7.4 BM DC Purification... 37
7.5 T cell purification... 38
7.6 Cell Lysate Preparation... 40
7.7 Co-culture A ssay... 40
7.8 In-vivo A ssay ... 40
7.9 Im m une M edia and Buffers ... 41
1. Introduction
1.1 Potential for Cancer Vaccines
Therapeutic cancer vaccines have advantages over traditional cancer treatments such as radiation and chemotherapy by having reduced side effects, and have the potential to be curative (1). Cell-based cancer vaccines hold great potential as the future of cancer therapy, and in 2010, Provenge (known generically as sipuleucel-T) became the first cell based immunotherapy approved by the FDA for treatment of advanced stage prostate cancer (2).
Cancer vaccines seek to target an antigen specific to a cancerous tumor and distinct from self-proteins; side effect occur when T cells target self proteins. An antigen may originate from the external environment or from within the body. Foreign, or non-self antigens, can activate a host immune system and trigger an immune response. By stimulating an immune response specific to the target antigens over-expressed by the cancer, it is the hope that a patient's immune system could be programmed to combat the cancer cells. There are several challenges which must be addressed for the development of a vaccine. A tumor can be comprised of many different cell types, all displaying multiple or different cell-surface antigens. Selection of an antigenic target which is expressed by the tumor cells and foreign to the individual proves to be difficult (3). Furthermore, cancer cells can mutate constantly, increasing the difficulty of identifying an appropriate target antigen (3). Another challenge which must be addressed is delivering the antigenic target to antigen presenting cells (2). Antigen presenting cells are a focus of cancer vaccine research because they hold great potential for activating the patient's immune system and to initiate a CD8+ T cell response. A cytotoxic T cell, also referred to as a CD8+ T cell or killer T cell, is a type of white blood cell that kills cancer cells, infected cells, or otherwise damaged cells. Most CD8+ T cells express T cell receptors (TCRs) that bind to specific antigens.
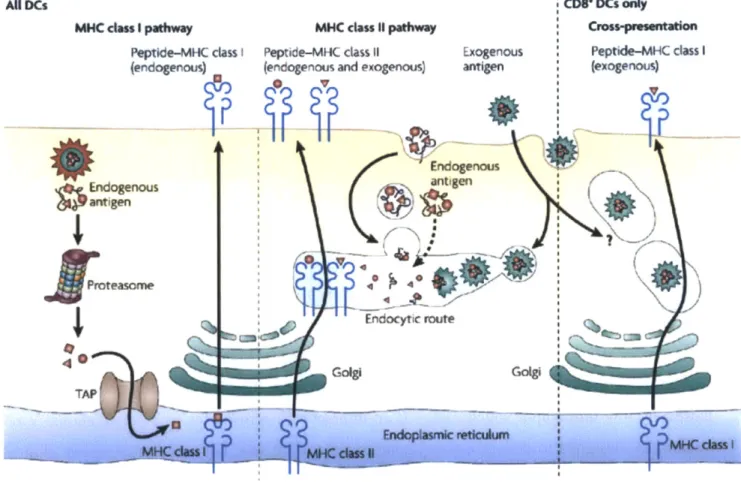
Antigens are presented to cells in the immune system via a histocompatibility molecule, which can then activate certain types of immune cells, such as T cells. For T cell receptor recognition, the antigen must be processed by the cell into small fragments and then presented to a T cell receptor via a major histocompatibility complex (MHC) (4). Each MHC molecule can display a molecular fraction of a protein, called an epitope. There are three subclasses of MHC molecules, class I, class II, and class III. Attention in research is typically given to class I and class II MHC
molecules. MHC II regulates specific immunity, also known as acquired immunity, to a specific antigen, and MHC Class I executes the destruction of host cells which have surface markers displaying that antigen. Experimentally, a CD8 effector response differs from a CD4 effector response. When proteins are found in the cell cytosol and then displayed on extracellular MHC Class I, a CD8 response is induced. When extracellular proteins are captured by endocytosis and displayed on MHC Class II markers, a CD4 response is induced. A phenomena known as cross-presentation, when endocytosed antigens are displayed on MHC Class I as opposed to MHC Class II has been observed. The mechanism underlying cross-presentation is poorly understood however, and is an inefficient method to target MHC Class I presentation (5). One of the greatest challenges in the development of these vaccines is creating antigen presentation by delivery of antigens into the cell cytoplasm to initiate a CD8 response as opposed to an endocytotic pathway, which would induce a CD4 response.
AU DCs CDS DCs only MHC dss I pahway Peptide-MHC c (endogenous) 4 Endogenous antigen 1 roteasome TAP
MWC cass 11 pathway Crimis-prwaton
ass i Peptide-MHC class I Exogenous Peptide-MHC class I (endogenous and exogenous) antigen (exogenous)
Endogenous
antigen
Endocytic route
G01191 Golgi
II
Nature ReviewsI Immunology Figure 1. MHC Class I and MHC Class II Pathways. Antigens can enter cells in a variety of
peptide is displayed on MHC Class I. When an antigen enters the cell via a endocytotic route, the antigen is displayed on MHC Class II.
1.2 Current Methods of Antigen Delivery and Their Shortcomings
The cell membrane is typically impermeable to macromolecules, and intracellular delivery of macromolecules is a significant therapeutic and research challenge. Existing techniques include nanoparticles, cell penetrating peptides (CPPs), electroporation, microinjection, sonoporation, and viral methods (6). In nanoparticle based methods, the target material forms a complex with a carrier material and is endocytosed by the cells of interest. Once inside the cell, the target material exits the endosome using the endosome escape mechanism to enter the cell cytoplasm (7). While and promising for in-vivo delivery environments, nanoparticles have significant potential for off-target effects and toxicity depending on the carrier composition. Current literature also has shown limitations to primary cell delivery and target materials other than nucleic acids (8). Cell penetrating peptides methods involve using a class of polypeptide sequences bound to target material that is subsequently endocytosed by the target cell, and is released from cellular endosomes to the cell cytoplasm (6). Cell penetrating peptides are capable of delivering a variety of materials, but much of the delivered material remains inactive in endosomes. In electroporation, cells are submitted to an electric field in the presence of target material, which can then diffuse into the cell cytoplasm through temporary pores created by the electric field. Limitations for this method include high toxicity and limitation of target material. Sonoporation exposes cells to an ultrasound which causes membrane disruptions, allowing the target material to enter the cells; sonoporation has similar drawbacks to electroporation. In microinjection, cells are penetrated with a microneedle and the target material is directly injected into the cell cytoplasm. While this method is theoretically capable of delivery of any target material to any cell type, the method is time consuming and extremely low throughput which makes it an impractical approach for most applications. Viral vectors are designed to deliver a target gene to cells of interest, but face limitations in clinical applications.
1.3 Rationale for New Intracellular Delivery Platform
The use of a new method of intracellular delivery, "squeezing," based on a microfluidic platform, will be explored for delivery to immune cells (6). The "squeezing hypothesis" states that the rapid deformation of a cell, as it passes through a constriction smaller than the cellular diameter, results in transient membrane disruptions as a result of the shear and compressive forces the cell experiences. These forces result in transient "pores" or "holes" in the lipid bilayer of the cellular membrane, allowing for material in the surrounding buffer to passively diffuse into the cell cytoplasm during the time span that the holes exists. Based on this hypothesis, any material in the surrounding buffer that can fit through these pores should be able to enter the cell cytoplasm. The size of the pores was hypothesized to be a function of the shear and compressive forces experienced. This hypothesis was tested first on HeLa and then across a wider variety of cell types. Microfluidic devices with different design parameters (ie: varied length and width) were developed for use across cell lines.
Shear-based and scrape loading delivery methods which temporarily injure/disrupt the cell membrane allowing for passive diffusion of the target material have been previously explored (9). Advantages of these methods include high cell throughput but are generally not used because of low-viability and inconsistent reproducibility in the applied shear forces. This microfluidic squeezing platform attempts to address the shortcomings of existing shear-based delivery methods by creating a controlled physical environment which allows cells to be sheared in a reproducible manner. The microfluidic chip uses a pressurized system to pass cells, buffer, and delivery material through parallel channels containing a constriction. This microfluidic approach to delivery causes rapid mechanical deformation to cells as the cells are passed through a constriction 30-80% smaller than the cell diameter. The shear forces create transient pores in the cells which allow for diffusion of material from the surrounding buffer to enter the cytosol. The method has the advantage of being high-throughput and is largely not specific to target material or cell type, making it useful for many applications. Unlike the existing methods described above, it also does not rely on electric fields, exogenous materials, endocytosis, or chemical alterations of the target material.
With this microfluidic approach, one could envision a novel therapy by which immune cells are isolated from a patient's blood, treated with the device ex-vivo by delivering tumor
lysate from a biopsy, and then reintroduced to the patient to create a long-lasting, patient specific CD8 response.
2. CellSqueeze Platform Development
2.1 System and Device DesignEach microfluidic chip contains 75 identical parallel channels etched in silicone and sealed with a Pyrex layer. Each channel contains at least one constriction, and constriction dimensions currently vary from 4 to 9 um in width and 10 to 50um in length. The notation for identifying chips is given as shown in Figure 2.
Nomenclature: AA
-
(B) X C
C
AA
0-*
B
AA: Width of Constriction
B: Length of Constriction
C: Constriction Channel Repitions
Figure 2. Notation for Chips. The notation is described by the length of the constriction, the width of the constriction, and the number of constrictions in a chip. For chips with repeating channels, the length and width parameters for each channel are consistent throughout the chip. For example, a chip designated as 30-6x5 would have a constriction length of 30um, a constriction width of 6um, and each channel have 5 constrictions.
The channel funnels into and out of the constriction at a 15-degree angle. Since the microfluidic channels are symmetrical flow through each chip is allowed in either direction until the chip
clogs. Typically 2-20 million cells can be run through a device prior to clogging, with a throughput of 50,000-500,000 cells/second. Parallel channels were designed to both increase throughput and to ensure that neighboring channels could still operate under original experimental conditions after some channels had clogged because the pressure system maintains constant pressure across all channels even if some clog.
/ /
/
/ 1~ / / / //
a) L#ore Weamn Inlet Flow MacromolcAs::Aftwr repid defornwtlOn
+- - utlet Delivred calls
0*
*00
le*
Figure 3. Chip/Holder Design and Delivery Mechanism. a) The delivery mechanism is believed to be one of passive diffusion of the target material into the cell cytol after experiencing shear and constriction forces as the cells pass through the squeezes of the chip. b) Image of the chip (2mm scale bar for size). c) Cells and their target material are mixed and then loaded into the inlet reservoir and can be collected from the outlet reservoir.
C)
Figure 4. Holder Set. The holder in its current iteration is made of injection molded ABS plastic. The inlet and outlet reservoirs are made of polycarbonate. The inlet and outlet reservoirs interface with the chip via O-rings.
2.2 Using the System
The chip is placed into a base-holder injection molded ABS plastic and is then interfaced with two identical reservoirs. O-rings are used to seal the connection between the reservoirs and the entry and exit ports of the chip. First, one reservoir is then loaded with the treatment cell population and target delivery material. The filled reservoir is attached to Teflon tubing, which is connected to a pressure regulator connected to a compressed air tank. The pressure regulator can be adjusted to operating pressures between 10-120psi, with the current design, and the pressure applied to the loaded reservoir drives the cells and delivery material through the chip and into the unloaded (outlet) reservoir. The sample can then be collected for further processing. Since the system design is symmetrical, the next sample can then be loaded into what was previously the "outlet" reservoir, and the process can be repeated until clogging occurs. To avoid contamination of cell samples, target delivery materials, and different delivery concentrations, chips are discarded and replaced with new chips when switching between cell types and target delivery materials. Additionally, for dosage dependent studies, concentration of the dosage was run experimentally in increasing order.
2.3 Efficacy of the Device on HeLa Cells
In order to evaluate the microfluidic platform and its potential for intracellular delivery, the hypothesis was first tested by attempting to delivery fluorescent dyes of varying sizes to HeLa cells. HeLa cells are a commonly used cell line, making it a good first candidate to test the system on. Fluorescent dyes were chosen as a preliminary delivery material because they are easy to use and their availability in different sizes would serve as a good preliminary method to determine the approximate pore size that could be formed. (eg: if delivering 3kDa, 70kDa, and 200kDa dyes and analysis showed that delivery of 3kDa and 70kDa was successful but that of 200kDa was not, one could assume that max pore size lay between 70-200kDa). Their fluorescent properties also allowed for read-out by flow cytometry.
2.3.1 Facs Data Analysis
Results for fluorescent dye assays were measured by flow cytometry, and "proof-of concept" dye deliveries typically measured 70 kDa fluorescein labeled Dextran and 3 kDa cascade Blue labeled dextran as the target material. Delivery efficiency by the device was compared in each experiment to cell samples that had not been exposed to the target material ("No Contact" or NC) as a negative control and to account for any autofluorescence of the sample. Samples treated by the device were also compared to samples in the same delivery solution that had been exposed to the target delivery material at the same concentration and for at least the same amount of time as the treatment cases as a comparison to endocytosis and surface binding of the target material by the cells ("Inlet" or IN).
2.3.2 Defining Delivery Efficiency
In order to measure the delivery level above that exhibited by autofluorescence, endocytosis, and surface binding, the delivered region is gated when measured by flow cytometry such that only the upper 1-5% of cells in the control and/or endocytosis sample fall into it. Treatment by the device does not affect autofluorescence levels of HeLa cells or immune cells (6).
0 1 1 wo Pacifi Bkj 60 40 fto- 0-j~4 1o~
Figure 5. Defining Delivery Efficiency. This illustrates typical gating for readout by flow cytometry. The x-axis represents fluorescence intensity and the y-axis measures number of events. a) illustrates the typical output for a no contact or endocytosis case. The upper 1-5% of viable cells are used to determine the gate cut-off. b) Typical output for a device treated case.
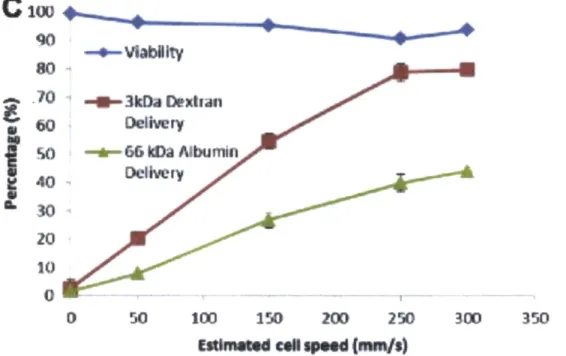
2.4 Assessment of Potential for Protein Delivery
The potential to deliver proteins to the cytoplasm of cells was assessed by delivery of fluorescein labeled albumin and dyes that were on a similar size scale to common proteins. HeLa cells were used in preliminary assessments.
C
IO 80 -- 70 -s-3kDa [*xtanoo
DeliveryI
'O -*--66 Wa Albumin 40 S30 20 10 0 0 50 100 150 200 250 300 350EtiKoEd col speed (nun/s)
Figure 6. Viability and Delivery for HeLa Cells at Varying Delivery speeds. 66kDa Albumin and 3kDa cascade blue dextran was delivered to HeLa cells at varying speeds. Delivery efficiency increased with delivery speed, and viability was assessed with propidium iodine stain.
2.5 SEM Studies
It was hypothesized that the membrane disruptions caused by the chips created temporary "pores" or holes in the external surface of the cell through which target delivery material could diffuse into the cytoplasm. Scanning electron microscopy studies were conducted in an attempt to visualize these physical changes. High resolution SEM images [JEOL 5910 General Purpose SEM] of gold coated HeLa cell samples before and after treatment by the device were produced to study any topological changes and to attempt to visualizes pore formation. See Appendix 7.1.
In order to capture the state of the cell membrane right after passing through the chip, a fixation solution was placed in the outlet reservoir of the device. Control cases included fixation of untreated cells, and fixation of cells 20 minutes after delivery.
Untreated Control cells bumpy/creased surface
Cells after saueezing smooth surface
Cells 20min after squeezing Return to normal morphology
Figure 7. SEM Images of Fixed Cells Under Various Treatment Conditions. The untreated cells had a rough and bumpy surface. The cells that were fixed immediately after treatment with the device were much smoother in comparison. Dark spots could be pores on the cell surface, but it was inconclusive as to whether they were actually pores or remains of some of the original bumps in the cell surface. Cells that were fixed 20 minutes after delivery have a topological appearance that lies somewhere between the treated and untreated case. There is a clear return of the bumpy surface structure, but the surface creases are not as defined or as abundant as in the untreated case. This suggests that the cells were recovering from the device treatment; these observations occur on a similar time scale to other experimental methods to determine pore closure time. The return to original cell morphology also supports experimental observations that the cells return to a fully functional state post-treatment by the device.
3. Using the CellSqueeze Platform to Enhance Antigen Presentation
for Cancer Vaccination Development
3.1 Assessment of the Delivery Potential of the CellSqueeze Platform to Primary Immune Cells
We targeted dendritic cells as attractive to inducing a cellular based immune response. The most powerful, antigen-presenting cells are mature, immunologically competent dendritic cells. Dendritic cells originate from hematopoietic stem cells in bone marrow and are distributed in their immature state throughout the body. During an immune response, immature dendritic cells are drawn to sites of inflammation and infection where they can internalize foreign antigens. The foreign antigens can then trigger the maturation of the DCs and trigger an immune response. Once the foreign antigens have been internalized and processed for display on MHC Class I and II, mature dendritic cells migrate towards the lymph organs which are rich in T cells. Mature dendritic cells can then promote T cell activation.
In these experiments, two types of dendritic cells were used--murine derived splenic dendritic cells and murine derived bone marrow dendritic cells (BMDCs). Response to treatment by the device in both systems illustrates that the treatment is applicable to both cell types and is not due to a characteristic artifact found in either cell type. Additionally, splenic and bone marrow derived cells have varying clinical and research implications. Bone marrow derived dendritic cells are more relevant to clinical applications because they are representative of monocytes which can be harvested from blood, and would be more relevant to clinical applications by having translational potential. In human based therapies, one could envision one day deriving monocytes from a patient's blood, differentiating the cells into dendritic cells, treating the cells ex-vivo, and re-introducing them into the patient as a personalized therapy.
3.1.1 Delivery of Dyes to Primary Murine derived Dendritic Cells (Spleen)
In order to investigate whether the CellSqueeze delivery technique could be used for primary murine derived immune cells, preliminary experiments were conducted to deliver 3kDa and 70kDa fluorescent Dextran to dendritic cells isolated from the spleen of B6 mice. 3kDa and 70kDa Dextrans were chosen because their size is similar to typical siRNA and protein molecules, respectively; dextran delivery is also easy to quantify by flow cytometry and dextran
molecules have minimal surface binding effects since they are negatively charged like the cell membrane.
3.2 Inducing Antigen Presentation In-vitro
In our preliminary assays in delivering target antigens to dendritic cells, we observed whether treatment of dendritic cells by the device could induce increased antigen presentation in vitro. OVA protein was delivered to the cell as a model antigen, and presentation of SIINFEKL on MHC Class I was measured by antibody staining. Upregulation of maturation markers CD80 and CD86 were also measured by antibody staining.
3.2.1 OVA as a Model Antigen
Ovalbumin (OVA), a major protein component of chicken egg whites, is an antigen commonly used as a model protein for the study of antigen-specific immune responses in mice. The OVA structure contains 385 amino acid residues including four cysteine residues and one cystine disulfide bridge (11). The signal sequence is in the middle of the polypeptide chain between residues 234 and 252. (12). The murine immune response is well characterized, and is often used in immunological assays. The MHC class I-restricted TCR transgenic line specific for ovalbumin has been generated (OTI), and will be used in our model OVA system assays.
3.2.2 OVA Delivery to Primary Murine Derived Dendritic Cells (Spleen)
To confirm that OVA peptide could be delivered to the cytoplasm of primary murine derived (C57BL/6) splenic dendritic cells, fluorescein labeled ovalbumin (OVA) was co-delivered with 3kDa blue dextran. Delivery efficiency of OVA, 3 kDa blue dextran, and propidium iodide were detected on the FITC, Pac-Blue, and PerCp channels respectively. As shown in Figure X, delivery of OVA to primary dendritic cells was comparable to delivery of the 3kDa blue dextran. Delivery efficiency increased with speed and viability dropped below 75%o only at >600mm/s. These assays helped characterize delivery conditions, such as speed through which the cells were run through the device and chip design type, at which subsequent experiments were conducted.
100%-
75%--
25%--.- 3kDa Dextran
0%/-
Ovalbumin
-A
-
Viability
0
200
400
600
800
Speed (mm/s)
Figure 8. Assessment of Delivery Potential of Dendritic Cells. In order to verify the delivery potential of dendritic cells, splenic dendritic cells were co-delivered with fluorescein labeled OVA and 3kDa cascade blue dextran. These results indicate that treatment by the device was capable of delivering both 3 kDa Dextran and OVA to primary dendritic cells. Additionally, increasing the speeds at which treated cells passed through the device increased the delivery of both 3kDa dextran and OVA up to 600mm/s. Viability was assessed by propidium iodine staining which marked dead cells.
3.2.3 OVA Delivery to Bone Marrow Derived Dendritic Cells (BMDCs)
Delivery potential of the OVA protein was evaluated for bone marrow derived dendritic cells (BMDCs) and splenic dendritic cells by measuring presence of SIINFEKL on MHC Class I by antibody staining. Efficacy of the system was illustrated by measuring MHC Class I presentation of SIINFEKL in both device treated and endocytosis cases. Since treatment by the device should have introduced OVA into the cell cytoplasm, MHC Class I presentation of the SIINFECKL peptide is expected. For endocytosis cases, less MHC Class I presentation is expected, because
endocytosis should lead to presentation on MHC Class 1I. Some cross-presentation may be expected. 14 I..., 105
013
08
MB
EB
SampIe
Siinfeki peptideUntreated cells
Device+Ova
Ova EndocytosisMHC-1 Stain
Spleenic
DCs
(l5hrs
post delivery)
'U 100- 80- 60- 40-
20-A
(k
0 0 02 10I
I
Bone Marrow Derived DCs (40-45hrs
Post
delivery)
100-80- g}Device
me ~60M}
Endo
'
40-20 0-/
o 102 3 4 105MHC-1 Stain
Figure 9. Antigen presentation in dendritic cells. Antigen presentation in a) splenic derived dendritic cells and b) bone marrow derived dendritic cells was assessed by delivering OVA as a model antigen and subsequently measuring the MHC Class I presentation of SIINFECKL via antibody staining 15 hours and 45 hours post-delivery, respectively. In both cases, treatment by
the device showed increased MHC Class I presentation when compared to endocytosis cases. Only viable cells were included and dead cells were excluded with propidium iodine staining.
3.3 Assessment of Normal Cell Function after Treatment by the Device
It was important to evaluate whether treatment by squeezing had negative affects on normal cell function post-delivery. This is particularly relevant to immune cells because the most potent antigen-presenting cells are mature, immunologically competent dendritic cells. Dendritic cells must convert from immature cells (before treatment) to mature, antigen-presenting, t-cell-priming cells which convey antigen presentation and express cytokines, chemokines, and other molecules which can initiate an immune response.
DC Progenitor Mature DC High MHC 1I, C180, CD86, CD40, CD25. IL-12, CD83, p55, Low FcR. CD54, CD58
4-Immature DC High intracellular MNC 11. FcR, Low C024, CD80, CD86. CD40. CD25. IL-12. CD83. p55
Figure 10. Dendritic Cell Maturation. Immature dendritic cells can become antigen-presenting mature DCs with the introduction of stimulatory factors such as LPS. Maturation can be measured by surface markers such as CD80 & CD86, abundant on mature DCs and low on immature dendritic cells.
3.3.1 DC Maturation Studies
Maturation studies were conducted to confirm that device treated dendritic cells would mature normally. OVA was delivered as the target antigen to primary dendritic cells. MHC Class I presentation of SIINFEKL was measured via antibody staining. Maturation of the treated cells was also measured by staining for upregulation of CD80 and CD86 in cells treated by the device, as mature cells are high in CD80 and CD86 while immature cells are low in CD80 and CD86. Maturation in treated cases was compared to maturation induced by lipopolysaccharide (LPS). Delivery of Ova and upregulation of CD80 and CD86 was measured in both splenic and bone marrow derived dendritic cells. The studies also showed that treated cells matured normally in comparison to LPS induced maturation cells.
100 so x 60 0 *40 20 0 102 0 10 4 , 5 0 102 10 3 ,0 1,
Antibody Stain against CD80 Antibody stain against CD86
Figure 11. Assessment of Maturation and normal cell function after treatment by the device. CD80 and CD86 upregulation was measured via antibody staining in order to assess whether treatment by the device would affect maturation. The untreated 4C condition was kept on ice for 3 hours in order to suppress all active cell processes as a negative control, while device treated cases and endocytosis cases were incubated at 37C with LPS, a maturation-inducing factor. Each treated device case and its corresponding endocytosis case have similar levels of CD80 and CD86 expression, indicating that treatment by the device does not cause the cells to rapidly endocytosis surrounding particles. This also indicates that treatment by the device does not inhibit the ability for cells to mature. The data shows that cells treated by the device show similar maturation as their untreated counterparts. Interestingly, the Untreated case which was not incubated with LPS also had a shift in both CD80 and CD86 expression compared to the 4C control, suggesting that the isolation process alone could also induce maturation as well. Results were measured using flow cytometry and dead cells were eliminated from analysis using propidium iodine stain.
3.3.2 Effect of Device Treatment on MHC I and MHC II Pathways
A novel advantage of the CellSqueeze platform is the potential to control the extent of the CD4 response. As described above, a CD8 response is activated through the MHC I pathway from antigens displayed in the cytoplasm and the CD4 response is activated through the MHC I pathway from antigens that have been endocytosed. We hypothesize that by varying the exposure to the delivery solution and antigen after treatment by the device, we can control the amount of
Soma" Untreated 4C - Endocytosis El Device+LPS E U untreated+LPS E Endocytosas*LPS
Longer exposure times will result in increased endocytosis, and short or essentially no exposure times (by washing the cells with PBS after delivery) will result in little or no endocytosis. Thus, long exposure times after treatment by the device would result in both a CD8 and CD4 response and shorter exposure times would lead to a CD8 exclusive response.
100 1ow 1 CA1 am-~ Lft 24-4 474 P5MCL-S 80
it~
~NWWUP 2. 11416 W-3lo* LPS -* *1 7 v 7I
60.1 !mLMS W32 m24i 20 o 1 10 5Figure 12: Treatment by the Device does not interfere with the CD4 pathway. CD4+ T cells were isolated from OT-I transgenic mice and co-cultured with BMDCs treated by the device. The experimental conditions (ex: 50:30) indicated the ratio of delivery buffer, typically DC Media containing cells at 5E6cells/mL to treatment solutions (1mg/mL OVA protein suspended in PBS). Since the proliferation between the 50:30 and 50:10 device treated conditions showed similar proliferation results as their respective endocytosis (inlet) cases while cells which did not come in contact with OVA but were treated by the device (no OVA) and untreated cells (NC,'no contact') showed no non-specific proliferation. These results were observed for cells that were incubated with and without LPS, a maturation inducing factor. The cells were measured on day 5 after treatment by flow cytometry by measuring CSFE staining.
4. In-vitro Assays for activating an Immune Response for a known
Antigen
The next step in testing the functionality of dendritic cells treated with the device was to determine whether treated cells could induce a T cell response in-vitro. To conduct these
experiments, we used OT-1 transgenic mice which are genetically altered so all their T cells have a T cell surface receptor for the SIINFEKL peptide. When their T cells are in the presence of a dendritic cell with the SIINFEKL peptide present on the MHC Class I, the T cell from the OT-1 mouse will bind to the dendritic cell and transition from its naive to activated state. The activated T cells initiate an immune response and can then proliferate. Measuring the extent of proliferation of the OT-1 T cells gives insight into the sensitivity of our system and the extent to which we can cause proliferation.
4.1 Co-culture Model
In our in-vitro assays, dendritic cells were treated by the device and the target antigen, OVA, was delivered. On Day 0 immediately post delivery, the dendritic cells were allowed to incubate at room temperature in the delivery solution (containing the delivery buffer such as PBS or DC Media and OVA) for 5-10 minutes, which is on the time scale of pore closure (6), to allow for diffusion of the peptide into the cell cytoplasm. The treated cells were then washed 2x with PBS to minimize endocytotic events and incubated at 37C 2 hours after LPS was added. After 2 hours, 30,000 dendritic cells were counted for each condition and incubated for 18-24 hours in a 96 well plate to allow for antigen presentation on the MHC I markers. On Day 1, T cells were isolated from OT-1 transgenic mice from the spleen and lymph nodes and stained with CSFE, which would later be used to detect the cells. 200,000 T cells were added to each dendritic cell condition from Day 0. It was determined via experimentation that 200,000:30,000 was the optimal ratio for T cells to dendritic cells, due to a combination of overcrowding factors and needing a critical number of activated dendritic cells to induce a T cell response. The supernatant media was subsequently changed on Day 2, and T cell proliferation was quantified on Day 4 by flow cytometry.
4.2 FACS: Measuring T cell Activation
T cells in our OVA assays should proliferate if they encounter the SIINFEKL protein on dendritic cell MHC Class I. In order to quantify this phenomenon, we stained our isolated T cells with CSFE prior to adding them to the dendritic cells on Day 1. Since CSFE is passed on from parent to daughter cells, each daughter cell is approximately half as saturated in the stain as the original cell. Thus the proliferation extent can be measured and correlated by the intensity of the
The lower the CSFE intensity (displayed on the FITC-A channel), the more proliferation cycles the T cells have undergone. T cell activation was measured by comparing fluorescence intensity on the FITC-A channel between device treated, endocytosis control, no contact control, and positive control samples. Live, non-activated T cells at the time of data analysis would present on the right of the histogram (high FITC intensity) because they have the highest intensity stain since they never proliferated. T cells which have been activated and subsequently proliferated would present closer to the left of the x-axis (low FITC intensity) the stain has been distributed to daughter cells during each division cycle.
300 MB 0".1 LPS IN u NC LPS B SO0-1 LPS IN W10 LPSIN B 50-0.1 LPS 200 * 'L *W5101,5 100 0 102 3 4 0
Ae"a Fluor 489-A
Figure 13: Proliferation results from CSFE stained T cells from OT-I transgenic mice co-cultured with bone marrow derived dendritic cells (BMDCs) treated with OVA. BMDCs were treated with treated with 30-6iS device types and all conditions were incubated with LPS for 2 hours post delivery. Siin and NC represent the positive control case (cells incubated with the SIINFEKL peptide) and untreated case, respectively. Experimental conditions (50-10, 50-1, and 50-0.1) and their respective endocytosis (inlet, or IN) cases are given by the volume ratio of cell solution (5.0e6 cells/mL in PBS) to the delivery solution (0.01mg/mL Ova in PBS). Proliferation results were measured on Day 4 after stained T cells and dendritic cells were co-cultured. The proliferation of the device treated cases over endocytosis cases was observed in all cases and is demonstrated by the lower fluorescence intensity and the higher cell counts, indicating proliferation. Endocytotic proliferation was only observed in the highest dosing case
(50-10) and was not present at the lower dosages (50-1 and 50-0.1). These results suggest that optimal dosing may occur between 50-1 and 50-0.1 conditions. This graph is a representative result of these assays.
Our results indicate that dendritic cells treated by the device were able to induce a T cell response in-vitro. Through our dosing assays, we determined that 50-10 and 50-1 conditions consistently was capable of inducing T cell proliferation while 50-0.1 conditions sporadically was capable of inducing proliferation. Our studies also suggest that at higher dosages, such as 50-10, endocytosis of the OVA protein was capable of inducing some CD8 T cell proliferation through the cross-presentation mechanism discussed previously, but was consistently less effective than the device treated cases at the same dosage.
4.3 Using Tumor Lysate as an Antigenic Source
Current methods which induce antigen presentation largely require the antigen to be identified and purified prior to delivery (ex: gplOO for melanoma). Some preliminary research has indicated that tumor lysate can be a potential source for delivering antigens and activating an immune response, which would reduce or eliminate the need for prior antigen identification (10). However, the methods used in these studies relied on endocytosis and cross-presentation, which are unreliable and unpredictable which leads to limited success.
Using the device as a treatment method to deliver tumor lysate directly to cells could potentially allow for treatment against cancers currently with no known unique markers. Tumor lysate, which contains a medley of different antigens present in the tumor cells, would be delivered to dendritic cells, which would subsequently display multiple antigens on their MHC Class I. These antigens, comprising of both self and non-self antigens, can then activate T cells to initiate an immune response. Ideally, self-antigens will be recognized by the innate immune system and tolerated, while non-self antigens will be recognized as foreign and targeted.
4.3.1 Rationale & Advantages
If we are able to successfully deliver tumor lysate to the antigen presenting dendritic cells and activate a T cell response, we will have the advantage of potentially targeting unidentified and/or
delivery multiple, unknown antigens simultaneously to dendritic cells, as the tumor lysate will contain multiple antigens present in the tumor from which it was obtained.
4.3.2 B16 OVA Melanoma Lysate Model
B16ova lysate is derived from b16 OVA melanoma cells which can be grown in culture. The b16 OVA melanoma cells over express the ova peptide. Lysate is created by a series of freeze/thaw cycles with the cells in suspension (Appendix 7.6). The supernatant of the lysed cells is collected and then delivered as the antigenic source to dendritic cells.
4.3.3 Lysate Dosage Testing for System Sensitivity
In lysate dosage co-culture assays, one would be able to evaluate the sensitivity of the system for identifying and displaying OVA out of an antigenic medley by delivering it at various dilutions to dendritic cells. Lysate derived from b16 OVA melanoma cells would be used as a model lysate. The lysate should be delivered with a normal running buffer (DC Media or PBS) and added in various concentrations to the buffer. For example, an assay may have several dilutions that range from 1:1, 5:1, and 5:0.1 buffer to lysate ratio. The lysate to ratio buffer can be optimized to maximize immune response. The buffer to lysate mixture should then be delivered as the antigenic source to primary murine derived dendritic cells. Based on whether proliferation is observed at various buffer to lysate ratios, and quantifying the amount of OVA which is present in lysate, one can evaluate how sensitive our system is to creating an immune response to a specific peptide from a medley of antigens.
1500
@ne
4)Goo
0103 14 lg0
ACIAx Flkjor 41%n-A
Figure 14. Proliferation results for CSFE stained OT-1 CD8 T cells co-cultured with BMDCs treated with B16-OVA lysate. Cells treated by 30-6 device designs and B 16-OVA lysate was delivered as the antigenic source. Proliferation was measured on Day 4 after stained T cells and treated dendritic cells were co-cultured. The treatment ratio (50:10) indicates the volume ratio of cell suspension (5.0e6 cells/mL in PBS) to cell lysate solution. Proliferation was observed in the two device treated cases, indicating that dendritic cells primed with lysate from treatment by the device are capable of inducing a CD8 T cell response. The lysate endocytosis (50:10 in) and untreated (NC) case did not have a measurable live cell population since the T cells died as a result of a lack of stimulation. These assays also lead to the hypothesis that the lysate itself may have an inhibitory or toxic effect as the lower dosage (50:1) case had more proliferation than the higher dosage (50:10) condition.
4.3.4 Lysate Toxicity Hypothesis
Based on data from preliminary co-cultures in which b16 OVA lysate was delivered as the antigenic source to primary murine derived dendritic cells, it was determined the lysate may cause a inhibitory or toxic effect. This was hypothesized because of lack of proliferation or less proliferation in higher lysate to buffer delivery conditions while experimental conditions
containing a lower lysate concentration showed proliferation. In order to investigate this hypothesis further, an OVA spike condition can be conducted.
4.3.5 Lysate Toxicity Assay
In order to assess the whether delivering tumor lysate as an antigenic source resulted in dendritic cell toxicity (subsequently observed by lack of an immune response) a lysate toxicity assay was developed. In this assay, one delivers B16 melanoma lysate in varying dosages while delivering a constant dosage of OVA protein to primary, murine derived dendritic cells in the co-culture model described above. Following these experiments, one would be able to conclude that lysate had inhibitory or toxic effects if there was less proliferation observed in conditions that contained a higher B16 Melanoma lysate dosage. One could conclude that the lysate did not have inhibitory effects if proliferation measured across all lysate to buffer ratios was consistent.
4.3.6 Lysate Characterization
Currently, work has already begun with B16 OVA lysate as an antigenic source using the co-culture model and the in-vivo model. Future work will include characterizing lysate used in experiments which will allow for quantification of OVA levels present in our lysate using current preparation methods. This knowledge could aid in modifying the preparation protocol to optimize antigen levels or improve lysate toxicity assays by allowing us to know whether current lysate dosages deliver OVA at a comparable level as the preliminary OVA assays which were conducted.
5.
In-vivo Assays for Activating an Immune Response for a Known
Antigen
5.1 In-vivo Proliferation Translation Assay
To evaluate the translation potential of the co-culture model, in-vivo proliferation studies were conducted. In our in-vivo assays murine derived CD8 T cells were isolated and purified from OT1+Ly5.1 mice and CSFE stained prior to retro-orbital injections on Day 0. The Ly5.1 gene is used to identify the injected cells from native T cells in follow-up analysis by staining for CD45.1. On Day 1, dendritic cells from B16F1O were treated by the device and the target antigen (ie: OVA) was delivered. The treated dendritic cells were then injected into the footpads of the
mice. The immune response was quantified by measuring T cell proliferation of the CSFE stained injected cells 3 days later from the draining lymph nodes, the non-draining lymph nodes, and spleen. In future work, we will also assess the versatility of our assay and approach by demonstrating similar results using the PMEL-1 model.
NC 2 OLN
100
NC 1LN IN 2 OLN Sin 1 DLN Device 2 DLN 6040-
L
20
0-0
l0o14
0
Figure 15. Proliferation Results for CSFE stained OT-1 CD8 T cells injected into B16F10 mice and subsequently injected with BMDCs treated with OVA. T cells were isolated from OT-1 transgenic mice, stained with CSFE, and subsequently injected into B16F1O mice on Day 0 using retro-orbital injections. On Day 1, BMDCs were treated with 30-6 design device and injected into the footpads of the mice previously injected on Day 0. On day 4, T cells were isolated from the draining and non-draining lymph nodes of the injected mice. Experimental conditions (Device) and their respective endocytosis (inlet, or IN) cases are given by the volume ratio of cell solution (5.0e6 cells/mL in PBS) to the delivery solution (0.01mg/mL Ova in PBS). Device cases generally proliferated over the inlet conditions in the draining lymph nodes and little to no proliferation was detected in the non-draining lymph nodes.
6. Future work & Project Outlook
By working with the CellSqueeze microfluidic platform, we are creating new ways to manipulate the innate immune response to facilitate advances in immunotherapy. In this work, we have investigated the ability of the device to deliver target antigens, measured antigen presentation on MHC Class I, assessed for normal cell maturation, and assessed the ability of treated dendritic cells to initiate a CD8 immune response. We investigated the ability of the device to deliver antigens to both murine derived splenic and bone marrow derived cells and to initiate a CD8 immune response in-vitro. Preliminary studies have also shown promising results for using cell lysate as an antigenic source. Moving forward, lysate models will continue to be explored, as they hold potential for activating an immune response against unidentified targets.
In the future, we envision an approach where a patient's cancer can be treated by this system if their tumor is unresponsive to traditional treatments. A sample of the tumor could be obtained via biopsy and mechanically lysed. The patient's dendritic cells can be harvested from his blood, and treated with the tumor lysate, ex-vivo. The patient's dendritic cells will then display a variety of markers on their cell surface and can be reintroduced into the patient's blood. The treated dendritic cells will initiate an immune response from the variety of antigens present on their MHC Class I receptors. Ideally, self-antigens will be tolerated by the immune system while foreign antigens will initiate a CD8 T cell response. In this system, one can induce a targeted, patient-specific, specific response to the tumor without previously having identified a unique target. Additionally, this multipronged approach can potentially attack multiple targets simultaneously since the lysate contains multiple antigens.
7. Appendix & Methods
7.1 Preparation of SEM Samples
Hela cells were treated by the device as previously described (6). 1OOuL of glutaraldehyde (initial concentration 25% v/v) solution was used for fixation. For fixation of the device treated sample, 100uL glutaraldehyde was placed in the outlet, and for the untreated case and treatment 20 minutes after device case, 1OOuL gultaraldehyde was added to 500uL of the cell suspension. The cells were washed with PBS, re-suspended in 2mL of glutaraldehyde solution (5% v/v), and
incubated for 10 minutes at room temperature. The cells were then washed in ethanol with 30%, 50%, 70%, 95%, and 100% v/v alcohol in distilled water solution, respectively. The cells were kept in each alcohol solution for 20 minutes and the ethanol solution was removed by 5s centrifugation between each wash. 15uL of each sample was then pipetted onto silicon grids and left at room temperature overnight prior to gold coating by the imaging core staff.
7.2 Running the Chip
Each microfluidic chip (Fig. #) contains 75 parallel microfluidic channels, containing one or more constrictions of set dimensions etched in silicon and sealed with a Pyrex layer. Width and length of each constriction on the chips vary from 4-8um in width and 10-40um in length. The parallel channels increases throughput of treated cells. The primed cells are mixed with the desired delivery material and then placed into an inlet reservoir. Teflon tubing is then attached to the inlet. A pressure regulator is used to specify the pressure desired at the inlet reservoir and then drives the passage of the cells from the inlet reservoir to the outlet reservoir. The treated cells can then be collected from the outlet reservoir and plated on a 96-well plate. After a 5-10 min post-delivery incubation, additional media is added to each condition, and each experimental condition is washed with PBS and resuspended in media. The cells are then incubated for 18-24 hours to allow for presentation of the delivered materials on MHC Class I.
7.3 Bone Marrow Dendritic Cell Creation
3-5 B6 mice are used to generate bone-marrow dendritic cells. Undifferentiated cells are harvested from the femur and tibia of the B6 mice and cultured with media containing GMCSF
(Granulocyte-macrophage colony-stimulating factor), a growth factor which promotes
differentiation to dendritic cells. The differentiation requires the undifferentiated cells to be cultured for 6-8 days, after which the differentiated dendritic cells can be purified for experimentation.
BMDC Harvesting Protocol: Materials:
Razor blades (2) 66 Filters (1 white) 66 Insulin Syringe (1) 66
BMDC Media and DC Media at 25C
Procedure:
1. Kill mice (1 per flask) & prep (spray in, get paper towels, wipe tools with EtOh)
-Fill 2 wells in a 6 well plate with DC media (pour enough to cover bottom of petri dish) in Dissection Hood
2. Cut leg, peel skin back to foot, remove. For remaining skin on leg, peel up to lower back, fold over.
3. With mouse on its back, bend femur and feel for joint. Place razor blade in bend and wiggle femur until loose.
-Cut away extra muscle with scissors. Isolate femur and calf bone and put in petri dish of EtOH.
4. Repeat for other leg.
5. Repeat for each mouse, using a new razor blade each time and cleaning scissors/tweezers. 6. Remove mice from hood, clean hood. Change gloves!
7. Take petri dish with bones
-For each bone, dip into EtOh, cut away all remaining muscle so there is only bone left.
-Dip into EtOh. Cut off ends (knobby part) of bone with new razor blade, put into new petri dish with DC Media
-Repeat for all four bones so that your new petri dish has 4 bone segments in it 15. Load syringe with DC Media. Take each bone segment and rinse through 3x with the
needle/syringe/media into the petri dish containing the other bone segments/media -For lower bone, needle should go in small end.
-Place empty bone into EtOh lid. 16. Repeat for each bone.
18. Using 25mL pipet, pipet up the media containing bone marrow cells and pass through a white filter.
19. Pipet up the remaining media in petri dish and run through filter. 21. Spin down at 500rcf for 4 minutes
-Prepare X (1 per mouse) flasks with 30mL BMDC Media at 25C
22. Pour away supernatant after spindown.
23. Resuspend in 10mL BMDC Media per mouse and add 10 ml to each flask 24. Label each flask with name, date, BMDC cells
25. Incubate at 25C (This is day 0) 26. Add 30mL BMDC Media on 3rd day.
7.4 BMDC Purification
The cell flasks containing the BMDCs are inspected to ensure the adherent DCs are uniform in appearance throughout the flask. A cell scraper is used to remove the cells from the flask surface. The cells are then centrifuged and resuspended in dendritic cell media and incubated with cd 1ic, mouse beads for 20 minutes on ice. The cd1ic beads are positively select for BMDCs and tag them with a magnetic bead. Post-incubation, the cells are centrifuged and filtered through a 50um filter into a primed MACS LS column. The BMDCs are then ejected from the column with MACs buffer, centrifuged and then resuspended again in the delivery buffer.
Materials: 2 white filters Microbeads, mouse 1 large MACs column
1. Set up hood, take 2 flasks from incubator and cell scrapers. 2. Check cells to make sure there's no contamination
3. Using cell scraper, scrape flasks. Check under microscope to see if more is necessary. Scrape again if nec.
5. Repeat for other flask.
6. Spin down @500rcf for 4 min
7. Aspirate supernatant and resuspend in 500ul DC Media per 50mL falcon tube. 8. Put a white filter on a new 50mL falcon tube and pass cells through.
9. Add microbeads, mouse. lOOuL for 1 flask, 120uL-15OuL for two flasks, depending on size of pellet in each flask.
10. Incubate on ice for 20min 11. Spin down @500rcf for 4 min. 12. Prep column with 3mL MACs buffer
13. Resuspend cells after they have spun down in 3mL MACs buffer 14. Pass through white filter into column
15. Wash through column with 3mL aliquots MACs 3x 16. Fill column with 6mL MACs
17. Use syringe and collect cells in a new 15mL falcon tube. 18. Spin down, resuspend in delivery buffer.
19. If waiting for a long time before using, add 6mL DC Media to the falcon tube to preserve viability. 7.5 T cell purification Materials: 3 blue filters 5mL syringe CD8 kit or CD4 kit
1 large column per 2 mice Lymph nodes and spleens DC Media
MACS buffer
1. Place 1 filter on a 50mL falcon tube. Wet with 2mL DC Media. (There should be 2mL DC Media in the falcon tube now)
2. Transfer spleens and lymph nodes onto the filter. Using the plunger part of the syringe, mash spleens against filter.
3. Wash filter with DC Media until clean. CHANGE TIP EVERY TIME!! 4. Prepare for cell count:
a. Mix the DC Media with 1000ul pipetter
b. Mix lOul of the cell suspension with 90ul DC Media
c. Take lOul of mixed cells from b. and mix with lOul trypan blue d. Count
e. Determine total cell count
Spin down the original 50mL falcon tube of cells @500rcf for 4 min Aspirate supernatant
Resuspend in 4OuL MACS buffer for lx^OA7 cells, based on count from part 4e. Pass through white filter into new 50mL falcon tube
Add 1OuL antibodies from CD8 or CD4 kit per lx^OA7 cells
If calculations yield greater than 200uL for CD8 or 400uL for CD4, add 200ul or 400 ul, respectively.
Incubate on ice for 10 minutes
Add 30uL MACS buffer for lx^OA7 cells, based on count from part 4e. Add 2x the volume of beads as antibodies added in part 8.
Incubate on ice for 15 minutes. Spin down @800rcf for 5 min.
Prep 1 large column with 3mL MACS buffer
Aspirate supernatant after spindown from part 13 and resuspend cells in 3mL MACS buffer.
Pass cells through white filter into column Wash 3x with 3mL MACs each time.
Keep the flow through and spin down @500rcf for 4 min Repeat steps 15-18.