Epigenome confrontation triggers immediate
reprogramming of DNA methylation and transposon
silencing in Arabidopsis thaliana F1 epihybrids
Mélanie Rigala,b, Claude Beckerc, Thierry Pélissiera,b, Romain Pogorelcnika,b, Jane Devosc, Yoko Ikedaa,b,1, Detlef Weigelc,2, and Olivier Mathieua,b,2
aLaboratoire Génétique Reproduction et Développement, CNRS, UMR 6293, INSERM U1103, F-63001 Clermont-Ferrand, France;bLaboratoire Génétique Reproduction et Développement, Université Clermont Auvergne, Université Blaise Pascal, BP 10448, F-63000 Clermont-Ferrand, France; andcDepartment of Molecular Biology, Max Planck Institute for Developmental Biology, D-72076 Tuebingen, Germany
Contributed by Detlef Weigel, January 27, 2016 (sent for review August 19, 2015; reviewed by Steven E. Jacobsen) Genes and transposons can exist in variable DNA methylation
states, with potentially differential transcription. How these epialleles emerge is poorly understood. Here, we show that crossing an Arabidopsis thaliana plant with a hypomethylated genome and a normally methylated WT individual results, already in the F1 gen-eration, in widespread changes in DNA methylation and transcrip-tion patterns. Novel nonparental and heritable epialleles arise at many genic loci, including a locus that itself controls DNA methyl-ation patterns, but with most of the changes affecting pericentro-meric transposons. Although a subset of transposons show immediate resilencing, a large number display decreased DNA methylation, which is associated with de novo or enhanced transcriptional ac-tivation and can translate into transposon mobilization in the progeny. Our findings reveal that the combination of distinct epi-genomes can be viewed as an epigenomic shock, which is charac-terized by a round of epigenetic variation creating novel patterns of gene and TE regulation.
DNA methylation
|
transcription|
transposable elements|
gene silencing|
ArabidopsisI
n eukaryotic genomes, cytosine methylation represents an epi-genetic mark involved in the silencing of transposable elements (TEs), genes, and transgenes (1, 2). In plant genomes, TEs are typically silent and associated with dense DNA methylation in the three cytosine contexts CG, CHG, and CHH (where H is any base but G). Repression of gene transcription by DNA methylation often correlates with methylation of promoter sequences whereas tran-scriptionally active protein-coding genes tend to be methylated ex-clusively at CG positions in their bodies (3–6).In the plant Arabidopsis thaliana (Arabidopsis), faithful prop-agation of CG methylation patterns upon de novo DNA synthesis during DNA replication is safeguarded by the DNA methyl-transferase METHYLTRANSFERASE 1 (MET1), the plant homolog of human DNA methyltransferase 1, such that sym-metrical CG sites in the genome are usually either fully meth-ylated or not at all (7, 8). Maintenance of non-CG methylation is more complex and involves the partially redundant activities of the DNA methyltransferases DOMAINS REARRANGED METHYLTRANSFERASE 2 (DRM2), CHROMOMETHYLASE 2 (CMT2), and CHROMOMETHYLASE 3 (CMT3). The three proteins act in self-reinforcing methylation and silencing loops that also rely on histone H3 methylation at lysine 9 (H3K9me) and small RNAs of 24 nt in length (9–12). The interplay between chro-matin and methylation is also apparent from the activity of the DECREASE IN DNA METHYLATION 1 (DDM1) chromatin remodeler, which seems to control access of methyltransferases to their H1-containing heterochromatic DNA targets (10).
Similar to changes in DNA sequence, differences in DNA methylation, either of natural, spontaneous origin or experimen-tally induced, can impact genome stability, gene expression, and phenotypic variation. Deficiencies inDDM1 induce drastic hypo-methylation of heterochromatin at all cytosine contexts, resulting
in transcriptional up-regulation of some heterochromatic genes, a massive activation of TE transcription, and mobilization of various TEs (10, 13–18). Elimination of MET1 in null met1 mutants causes a near-complete loss of CG methylation at genes and TEs, which is associated with misexpression of numerous protein-coding genes and broad transcriptional activation of TEs (3–5, 8). However, although transcriptionally activated, some autonomous TEs remain immobile, owing to other si-lencing mechanisms; for example, transposition of the Evadé (EVD) retrotransposon occurs only after several generations of inbreeding or in combinations ofmet1 with mutations in other silencing factors (2, 19, 20).
In addition to the loss of CG methylation,met1 mutants dis-play substantial ectopic CHG and CHH hypermethylation at thousands of genes and TEs (4, 21, 22). Depending on the target, this aberrant methylation has been linked to erratic or mis-targeting of RNA-directed DNA methylation (RdDM), tran-scriptional silencing of the gene encoding the REPRESSOR OF SILENCING 1 (ROS1) DNA demethylase, and misregulation of
theINCREASE IN BONSAI METHYLATION 1 (IBM1) gene,
which encodes an H3K9me2 demethylase that specifically targets protein-coding genes (22–24). IBM1 contains a large intron with a heterochromatin-like segment that is methylated at CG and non-CG
Significance
Similar to changes in DNA sequence, induced or naturally oc-curring variation in cytosine methylation can impact gene expression. How distinct methylation states of genes and transposons, called epialleles, emerge is not well understood. Here, we report that combining identical genomes with dras-tically different DNA methylation patterns in the same indi-vidual results in an epigenomic shock that is characterized by widespread changes in DNA methylation and gene expression. Many novel epialleles not found in the parents are formed at genes whereas transposons often experience decreased DNA methylation associated with transcriptional activation. Our work provides a scenario for the rapid and broad-scale emer-gence of epigenetic variation and may have implications for transposon dynamics within populations.
Author contributions: M.R., C.B., D.W., and O.M. designed research; M.R., C.B., T.P., J.D., and Y.I. performed research; M.R., C.B., T.P., R.P., J.D., Y.I., D.W., and O.M. analyzed data; and M.R., C.B., D.W., and O.M. wrote the paper.
Reviewers included: S.E.J., University of California, Los Angeles. The authors declare no conflict of interest.
Data deposition: Sequencing data generated in this study have been deposited with the European Nucleotide Archive (ENA),www.ebi.ac.uk/ena(accession no.PRJEB9919). 1Present address: Institute of Plant Science and Resources, Okayama University, 2-20-1
Chuo, Kurashiki, 710-0046, Japan.
2To whom correspondence may be addressed. Email: weigel@tue.mpg.de or olivier.mathieu@ univ-bpclermont.fr.
This article contains supporting information online atwww.pnas.org/lookup/suppl/doi:10. 1073/pnas.1600672113/-/DCSupplemental. PLANT BIOLO GY PNAS PLUS
heterochromatin structure and is associated with impaired pro-duction of the longIBM1 transcript IBM1-L, which encodes the functional form of IBM1 (23, 25). In addition to the ROS1- and IBM1-mediated defects inmet1 mutants, genes premarked with H3K27me3 tend to gain ectopic H3K9me2 and to be depleted in H3K27me3 inmet1 whereas heterochromatic loci show deple-tion in H3K9me2 and ectopic H3K27me3 upon CG methyladeple-tion loss (26, 27).
In ddm1 and met1 mutants, changes in DNA methylation
generate epialleles without changes in DNA sequence. In a few examples, such epialleles have been associated with heritable differential transcription (7, 28–31). It has been long established that some ddm1- and met1-induced hypomethylated epialleles can be stably transmitted through several generations after out-crossing the causative mutation (13, 32). Similarly, hypermethylated epialleles can be transmitted across generations although they seem to be less stable than hypomethylated ones (29, 30, 33). To further analyze transgenerational epigenetic inheritance, segre-gation of allelic DNA methylation variants, or epialleles, was induced in epigenetic recombinant inbred lines (epiRILs) that were created by outcrossing either a met1 or a ddm1 hypo-methylated parent plant to an isogenic WT plant (34, 35). Studies of themet1- or ddm1-derived epiRIL populations have confirmed overall stable inheritance of parental DNA methyl-ation patterns but also have revealed some degree of epigenetic instability of parental epialleles. Inddm1-epiRILs, several TEs and single copy sequences inherited from the mutant parent undergo progressive DNA remethylation in a process requiring sRNA biogenesis (36) whereas, in later generations of some met1-epiRIL lines, nonparental methylation patterns can be detected, revealing the creation of further epigenetic novelty in these plants (34). Additionally, althoughEVD and CACTA TEs are immobile in both parents, proliferation of the two TEs was detected in a fraction of met1-derived epiRILs (19, 34), rein-forcing the notion that parental epigenetic regulatory circuits have been altered in these lines.
Here, we examined the immediate consequences of combining the contrasting WT and met1 methylomes in F1 plants. Using whole genome DNA methylation analysis in combination with sequencing of mRNA and small RNA transcriptomes, we reveal widespread nonadditive variation in DNA methylation, TE si-lencing, and gene expression. These effects translate into the
changes in silencing control and efficiency. Results
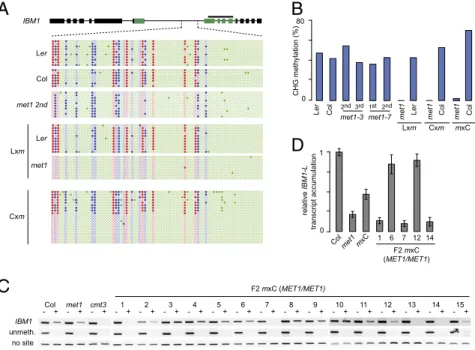
A Novel Nonparental Heritable HypomethylatedIBM1 Epiallele in F1 Epihybrids.We previously reported that properIBM1 transcrip-tion requires the concomitant presence of CG and CHG meth-ylation in the largest intron of the gene (23). To determine the role ofMET1 in methylation of IBM1 intron sequences, we crossed amet1-3 mutant (hereafter referred to as met1) in a Col-0 back-ground, which has reduced DNA methylation in this intron, to a Ler-0MET1 WT (MET1+) plant. Not only was CG methylation of themet1-derived IBM1 allele not restored in F1 plants (23) (Fig. 1A), but there was a further, drastic loss of intronic CHG methylation (Fig. 1A). This hypomethylation was not due to parental effects ofmet1 because a drastic loss of CHG methyl-ation was not seen upon selfing of met1 plants over several generations (Fig. 1B). These results indicated that the drastic loss of CHG methylation was a specific property of interaction betweenmet1 and MET1+genomes in F1 hybrid plants.
To eliminate effects caused by background genetic differences between the Col-0met1 and the Ler-0 MET1+parents, we out-crossed amet1 plant to an isogenic Col-0 MET1+plant. Because the genomes of these parents are nearly identical but differ drastically in DNA methylation patterns, we named the resulting F1 plants“epihybrids.” MET1+plants of the Col-0 and Ler-0 ac-cessions showed similar DNA methylation patterns at the IBM1 intronic region (Fig. 1 A and B). Independent of the ori-entation of the cross [Col-0 × met1 (C×m) or met1 × Col-0 (m×C)], we again observed the emergence of a nonparental DNA methylation pattern at DNA sequences that were assigned to the met1-derived IBM1 allele based on the absence of CG methylation (Fig. 1A and B). Similar results were obtained when we crossed themet1-7 allele in the Col-0 background to a MET1+ plant of the Van-0 accession (SI Appendix, Fig. S1A). Together, these results suggest that the appearance of the nonparental hypomethylatedIBM1 epiallele in F1 epihybrids is linked to the difference in DNA methylation patterns between parental plants. The mechanism involved in CHG methylation loss re-mains to be determined, but it seems to be independent of IBM1 and ROS1 (SI Appendix, Fig. S2).
To assess heritability of the novelIBM1 epiallele, we determined IBM1 intron methylation in F2 progeny of a self-fertilized m×C F1
IBM1 Col met1 2nd met1 Ler Ler Col Ler Lxm Cxm mxC met1 met1
met1 Ler Col Col
A
Col met1 cmt3 - + IBM1 - + - + unmeth. no site -1+ -2+ -3+ -4+ -5+ -6+ -7+ -8+ -9+ F2 mxC (MET1/MET1) -10+ -11+ -12+ -13+ -14+ -15+C
Col met1 evi tal er L-1 M BI noi tal u mu cc at pir cs na rt0 1D
mxC 1 7 12 14 F2 mxC (MET1/MET1) met1-3 ) %( noi tal yht e m G H C 0 80 met1-7B
2nd 3rd 1st 2nd Lxm Cxm 6Fig. 1. A nonparental IBM1 epiallele in F1 epihy-brids. (A) DNA methylation at the large IBM1 intron, as determined by Sanger sequencing of bisulfite-treated DNA. Filled and empty circles indicate methyl-ated and unmethylmethyl-ated cytosines, respectively; the color denotes sequence context (red, CG; blue, CHG; green, CHH). (B) CHG methylation of the large IBM1 intron, as determined by Sanger sequencing of bisulfite-treated DNA. Parental origin of IBM1 alleles in C×m and m×C F1 epihybrid plants was assigned based on presence/ absence of CG methylation. (C) DNA methylation at the large IBM1 intron, as determined by CHG methylation-sensitive AluI digestion followed by PCR. For each ge-notype, PCR amplification was performed on digested (+) and undigested DNA (−). A region unmethylated in the WT and a region deprived of the AluI site were amplified as controls of digestion efficiency and DNA loading, respectively. (D) Quantitative RT-PCR analy-sis of the accumulation of IBM1 full-length mRNA (IBM1-L) in the indicated genotypes.
individual. As expected for Mendelian segregation, one quarter of the MET1+F2 plants were homozygous for the hypomethylated IBM1 epiallele and accumulated only little IBM1-L transcript (Fig. 1 C and D). Additionally, absence of intronic methylation was inherited in F3 progeny of a self-fertilizedMET1+F2 individual homozygous for the hypomethylatedIBM1 epiallele (SI Appendix, Fig. S1B). Therefore, outcrossing a hypomethylatedmet1 parent to aMET1+plant induces the generation of a novel, nonparental hypomethylated IBM1 epiallele in F1 epihybrid progeny. The novel epiallele was stable through at least two generations, in-dependently of the presence of themet1 mutation.
DNA Methylation Variation in F1 Epihybrids Mainly Occurs at Pericentromeric TEs.Our observation at the IBM1 locus promp-ted us to examine the extent of DNA methylation variation in F1 epihybrid plants on a genome-wide scale. We performed whole-genome bisulfite sequencing in Col-0MET1+andmet1 parents and in pools of both reciprocal F1 epihybrids. We combined the parental datasets to reconstruct a synthetic F1 sample (hereafter named rF1) where each genomic cytosine is methylated at the midparent value (MPV). Overall DNA methylation levels were similar in the two reciprocal F1 epihybrids and showed a sub-stantial increase in CG methylation, decreased CHG methyl-ation, and localized changes in CHH methylation compared with rF1 (Fig. 2A). Gain of CG methylation was prominent at peri-centromeric regions and peaked at centromeres, where CHG and CHH methylation also showed the strongest deviation from rF1 levels (Fig. 2B). Consistently, TEs globally displayed in-creased CG methylation in F1 epihybrids relative to rF1 whereas CG gene-body methylation was at the expected rF1 level (SI Appendix, Fig. S3 A and B). These results indicate that most of the variation from the expected rF1 DNA methylation level occurs at pericentromeric regions of F1 epihybrids.
By comparing the two reciprocal F1 epihybrids to rF1, we determined differentially methylated regions (DMRs) at any of the three cytosine sequence contexts significantly deviating from the mere 1:1 addition of parental methylomes. We also
de-termined DMRs between the MET1+ and met1 parents. We
defined a total of 9,892 F1 hypomethylated DMRs (hypo-DMRs), where the F1 epihybrids had lost DNA methylation, and 8,892 F1 hypermethylated DMRs (hyper-DMRs), where the F1 epihybrids had gained methylation compared with the synthetic rF1 (SI Appendix, Fig. S3C). Independent of their type, the vast majority of DMRs matched TE loci, and gains in CG methyl-ation and losses in CHG methylmethyl-ation were the most abundant types of DMRs. In each case, more than 60% of the DMRs were common to reciprocal F1 epihybrids, indicating that most DNA methylation changes in epihybrids occur independently of cross
orientation, similar to the creation of the hypomethylated IBM1 epiallele.
We previously showed that methylation patterns are unstable upon met1 inbreeding (22). To rule out the possibility that methylation changes in F1 epihybrids merely reflect the contin-uation of alterations present in the met1 parent used, we also profiled DNA methylation in progeny of the self-fertilizedmet1 parent. The vast majority of the F1 DMRs did not differ between the two successive met1 generations (SI Appendix, Fig. S4), in-dicating specificity in the methylation changes occurring in F1 epihybrids. To further account for interindividual variation,
we profiled DNA methylation in five m×C F1 individual
plants. Most of the F1 DMRs were also detected in these plants (SI Appendix, Fig. S4). Finally, we determined DNA methylation patterns in met1/+ F2 progeny of self-fertilized m×C F1 epihybrids. F1 and F2 showed comparable methyl-ation at a large proportion of F1 non-CG DMRs (SI Appendix, Fig. S5A), indicating that methylation changes triggered in F1 epihybrids can be inherited in the next generation. In agreement with the requirement of MET1 for maintaining CG methylation during gametogenesis (8), CG methylation was reduced at pro-tein-coding genes and TEs in F2 compared with F1 epihybrids (SI Appendix, Fig. S5B). Noticeably, we observed gene-body CHG hypermethylation at IBM1 target genes in the F2 (SI Appendix, Fig. S5 B and C), further confirming segregation of the lowly expressed hypomethylatedIBM1 epiallele in F2 plants.
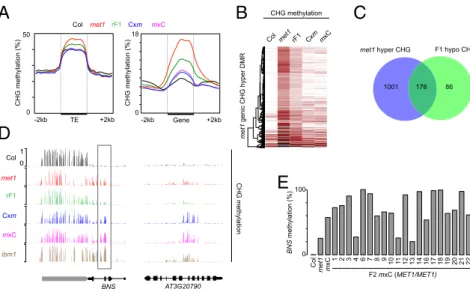
Ectopic CHG Methylation at Genes in F1 Epihybrids. Thousands of genomic loci are ectopically hypermethylated at CHG positions inmet1 mutants, in part due to misregulation of IBM1 and ROS1 (4, 22, 23, 26). Overall, genes, but not TEs, that are
CHG-hypermethylated in met1 continued to have a higher CHG
methylation level in F1 epihybrids compared withMET1+plants (Fig. 3A–C). Using published data (21), we defined genes ec-topically methylated at CHG positions inibm1 and in the ros1 dml2 dml3 (rdd) triple mutant, which lacks all three main DNA demethylases expressed in vegetative tissue. There was limited overlap between genes that are hypomethylated in F1, but hypermethylated inmet1 and ibm1 or in met1 and rdd, indicating that normal methylation at IBM1 targets and RDD targets was mostly not restored in the F1 (SI Appendix, Fig. S6A). We pre-viously demonstrated that restoring proper levels of functional IBM1 transcript accumulation in met1 resulted in removal of ectopic CHG methylation and H3K9me2 at selected genes (23). Also, we found thatIBM1 and ROS1 were transcribed at a near MPV in F1 epihybrids (Fig. 1D and SI Appendix, Fig. S6B). Together, these data suggest that the amount of functional IBM1 produced in F1 epihybrids is insufficient to immediately restore normal methylation, thus contributing to persistent ectopic CHG
A
B
0 3 −0.75 0 0.75 methylation difference density CG methylation Cxm-rF1 mxC-rF1 0 3 −0.75 0 0.75 methylation difference density CHG methylation 0 4 −0.75 0 0.75 methylation difference density CHH methylation -10 0 20 Chr 1 x C 1 F m 1 Fr -) %( ec n er effi d n oit al y ht e m b M 0 3 0 mCG mCHG mCHHFig. 2. Global DNA methylation changes in F1 epihybrids. (A) Kernel density plots of DNA methylation differences between C×m or m×C epihybrids and the reconstructed F1 (rF1) at CG, CHG, and CHH contexts. (B) Mean methylation difference between C×m and rF1 in 100-kb tiles along chromosome 1 (Chr1). The “m” prefix denotes methyl.
PLANT
BIOLO
GY
PNAS
hypermethylated inmet1, and not in either ibm1 or rdd, retained CHG methylation in F1 epihybrids (SI Appendix, Fig. S6A). Thus, whatever the cause of ectopic genic CHG hypermethylation in met1, this methylation largely persists in F1 plants.
We identified about 100 genes with CHG hypermethylation in each F1 epihybrid compared with either parent (SI Appendix, Fig. S7A). Although, at a small subset of genes, CHG hyper-methylation seemed to be initiated specifically in F1 plants (SI Appendix, Fig. S7 A and B), most transgressive CHG hy-per-DMRs already displayed a certain level of ectopic CHG methylation in themet1 parent (Fig. 3D andSI Appendix, Fig. S7A). About 60% of these genes were also CHG hyper-methylated in ibm1 (Fig. 3D andSI Appendix, Fig. S7C), sup-porting the notion that insufficient IBM1 activity is a major contributor to transgressive CHG hypermethylation in F1 epihy-brids. For example,BONSAI (BNS), the first identified IBM1 target (24), gained CHG methylation at the same positions inibm1 and, to a lesser extent, inmet1, and became further hypermethylated in F1 epihybrids (Fig. 3D). To determine which BNS allele was hyper-methylated in F1 epihybrids, we self-pollinatedm×C F1 plants, and assessed theBNS methylation state in MET1+F2 individuals. We
did not recover MET1+ F2 plants with MET1+-like BNS
methylation levels. In about three-fourths of the F2 progeny, the methylation level was similar or higher than in the F1 epihybrid parent whereas the other quarter showed amet1-like level of methylation (Fig. 3E). These results suggest that the MET1+-derived BNS allele acquired CHG hypermethylation in F1 epihybrids, creating novel nonparental BNS epialleles. Intriguingly, some F2 individuals with high BNS CHG meth-ylation levels had normalIBM1-L expression (Figs. 1D and 3E), suggesting that, unlike the original BNS allele in the MET1+ background, the novel CHG hypermethylated BNS epiallele formed in F1 epihybrids is largely insensitive to IBM1 activity. Similarly, ddm1-induced hypermethylated BNS epialleles are inherited after restoration of the DDM1 function (29).
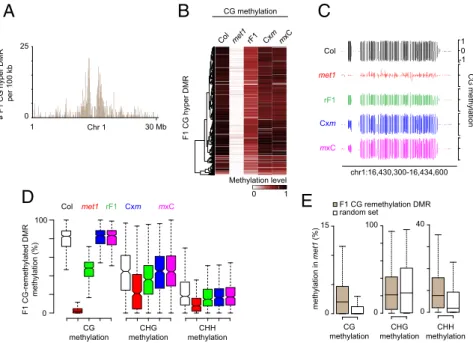
Immediate CG Remethylation at TEs in F1 Epihybrids.F1 CG hyper-DMRs mostly overlapped with TEs (2,138 TEs) (SI Appendix, Fig. S3C) preferentially located in pericentromeric regions (Fig. 4A). These genomic loci, which were highly CG-methylated in theMET1+plants, had lost most of their CG methylation inmet1 (Fig. 4B), indicating that F1 CG hyper-DMRs mostly reflect remethylation of the correspondingmet1-derived TE epialleles. At about 25% of remethylated loci, CG methylation was fully restored to theMET1+level in F1 epihybrids (Fig. 4C and D). The relative distribution of TEs overlapping with fully reme-thylated F1 CG hyper-DMRs closely mirrored the distribution of
showing that CG remethylation in F1 epihybrids affects all types of pericentromeric TEs. Notably, compared with an equivalent set of random TE regions, CG-remethylated loci retained more CHH methylation and a substantial CG methylation level in the nullmet1-3 mutant (Fig. 4E), indicating that part of CG meth-ylation at these TE sequences is independent of MET1 activity. Full restoration of CG methylation in F1 epihybrids was globally accompanied by reestablishment of CHG methylation whereas CHH methylation did not significantly deviate from the rF1 level (Fig. 4D).
We also observed CHH hyper-DMRs in F1 epihybrids al-though these DMRs were less common than CG hyper-DMRs (SI Appendix, Fig. S3C). CHH hyper-DMRs were enriched at the edges of pericentromeric LTR/Gypsy retroelements and were associated with DNA hypermethylation in all cytosine contexts, with transgressive CHH and CHG methylation where the F1 had higher methylation than either parent (SI Appendix, Fig. S8). This hypermethylation is likely mediated by DRM2, which preferentially targets the edges of long retrotransposons (10).
Transdemethylation of TEs in F1 Epihybrids. Hypomethylation in F1 epihybrids primarily affected TEs and was mostly due to changes at CHG and CHH positions, which often occurred
concurrently (SI Appendix, Figs. S3C and S9A). VANDAL
transposons were overrepresented among TEs with non-CG hypomethylation in F1 (SI Appendix, Fig. S9 B and C), which were characterized by very low DNA methylation in met1 mutants (Fig. 5A), indicating that DNA hypomethylation in F1 epihybrids affects TE copies inherited from theMET1+ parent.
A specificVANDAL21 element, Hi, transposes in ddm1 and met1 mutants (16, 18). The new Hi copies induce trans-demethylation, mediated by the Hi-encoded VANC protein, having a high specificity for VANDAL21 family elements and being more pronounced at non-CG sites (18). In our material,Hi was hypomethylated inmet1 at all cytosine contexts and asso-ciated with strong non-CG hypomethylation in F1 epihybrids (SI Appendix, Fig. S10A). We detected newHi copies in met1 and in F1 plants (SI Appendix, Fig. S10B), supporting that trans-posedHi copies may induce transdemethylation of VANDAL21 elements in F1 epihybrids (SI Appendix, Fig. S9C). Because many other elements from otherVANDAL families were also strongly hypomethylated in F1 epihybrids (SI Appendix, Fig. S9C), we sought to determine whether this hypomethylation was due to the same type of transposition-dependent transdemethylation mech-anism. We estimated TE genome-wide copy numbers by com-paring the methyl-C DNA sequencing coverage ofmet1 and F1
CHG methylation (%) 0 50 -2kb +2kb TE 0 18 -2kb +2kb Gene CHG methylation (%) Col met1 rF1 Cxm mxC
met1 hyper CHG F1 hypo CHG
176 86 1001 met1 genic CHG hyper DMR CHG methylation Col met1 rF1 CxmmxC CHG methylation AT3G20790 BNS Col met1 rF1 Cxm mxC ibm1 0 F2 mxC (MET1/MET1) 1 2 3 4 6 7 8 9 10 11 12 13 14 16 17 18 19 20 21 22 BNS methylation (%) 100 Col met1 mxC 1
A
B
D
C
E
0Fig. 3. Genic CHG methylation in epihybrids. (A) Average CHG methylation over TEs and genes containing at least one met1 CHG hypermethylation DMR. (B) Heat-map of CHG methylation levels within met1 CHG hypermethylation DMRs. Rows represent the DMRs, and the columns genotypes. (C) Venn di-agram of the overlap between genes containing at least one CHG hypermethylation DMR in met1 and genes containing at least one CHG hypomethylation DMR in F1 epihybrids. (D) Genome-browser views of CHG methylation at the indicated loci. (E) The DNA methylation level at the BNS locus in indicated gen-otypes was determined by McrBC digestion followed by qPCR. The individual m×C F2 plants analyzed in-clude the ones analyzed in Fig. 1 C and D.
epihybrid plants with that of theMET1+parents. Validating this approach, we detected an increase inEVD and Hi copy number in the second generation of themet1 homozygous line (SI Appendix, Fig. S10C). Additionally, we detected more DNA copies of the ATCOPIA21 element AT5TE65370 in met1, previously found to be active only in advanced generations ofddm1 mutants (16) (SI Appendix, Fig. S10C). Importantly, this analysis did not reveal mobilization of otherVANDAL elements in either met1 or F1 epihybrids (SI Appendix, Fig. S10D), suggesting that loss
of DNA methylation occurring at MET1+-derived VANDAL
elements in F1 epihybrids is neither necessarily caused by, nor associated with, transposition of TE members from the corre-sponding hypomethylated families. Considering the previously reported high specificity forHi-induced transdemethylation, we conclude that production of the VANC anti-silencing factor from transcriptionally activatedVANDAL elements in met1 is sufficient to induce transdemethylation of elements of the same family in F1 without requirement for transposition.
Transdemethylation Is Associated with TE Mobilization. The CG hypomethylation ofVANDAL elements was more pronounced in
their 3′ halves, which contain the ORF encoding VANC (Fig. 5A). F1 CG hypo-DMRs also overlapped with DNA/En-Spm DNA transposons (SI Appendix, Fig. S9B), and CG hypo-methylation was preferentially located near the borders of these elements (Fig. 5B). Studies of the maize Spm transposon have revealed that the transposon-encoded TnpA protein can in-duce DNA hypomethylation at TnpA binding sites that are located at the element ends, and promote its transcription (37, 38). The distribution of F1 CG hypo-DMRs suggests that a similar mechanism may operate at ATENSPM elements in F1
epihybrids. The En-Spm elementsCACTA1 (CAC1) and CAC2
transpose in ddm1 mutants and ddm1-epiRILs (14, 16, 35). CACTA movement has also been detected in met1-epiRILs al-though the mobilized elements were not assigned to a specific element (34). CAC1 is mobilized in met1 cmt3 but not in met1, indicating that both CG and CHG methylation cooperate to repressCAC1 activity (20). In F1 epihybrids, CAC1 edges were hypomethylated at CG sites and also displayed transgressive CHG hypomethylation (Fig. 5C). Because CG methylation was completely lost in the met1 parent, this result indicates hypo-methylation of theMET1+-derivedCAC1 allele in the epihybrids. Col met1 rF1 CxmmxC CG methylation F1 CG hyper DMR Methylation level 0 1
B
1 -1 0 Col met1 rF1 Cxm mxC CG methylationC
chr1:16,430,300-16,434,600A
# F1 CG hyper DMR per 100 kb 1 Chr 1 30 Mb 0 100 CG methylation CHG methylation CHH methylation F1 CG-remethylated DMR methylation (%) Col met1 rF1 Cxm mxCD
0 15 0 100 0 40 CG methylation CHG methylation CHH methylation methylation in met1 (%) F1 CG remethylation DMR random setE
0 25Fig. 4. CG remethylation in epihybrids. (A) Cumu-lative number of CG hypermethylation DMRs in F1 epihybrids in 100-kb tiles along chromosome 1. (B) Heat-map of CG methylation levels within CG hypermethylation DMRs in F1 epihybrids. (C) Genome-browser view of CG methylation at a representative genomic region displaying CG remethylation in F1 epi-hybrids. (D) Box plot of CG, CHG, and CHH methylation levels in F1 CG-remethylated DMRs. Values were extracted from one replicate per genotype. (E) Box plot of CG, CHG, and CHH methylation levels in F1 CG-remethylated DMRs and in a random set of sequences of equivalent type and length.
0 100 0 30 -2kb +2kb TE 0 50 -2kb +2kb TE 0 80 -2kb +2kb TE
CG methylation CHG methylation CHH methylation Col met1 rF1 Cxm mxC methylation (%)
E
A
CHG methylation (%) transgressive F1 CHG hypo-DMRs Col met1 rF1 Cxm mxC LTR/Gypsy F1 CHG hypo DMR density 0 1 5’ 3’ 0.4 -0.4 1 0 Col met1 Cxm - rF1 mxC - rF1 1 0 0.4 -0.4 0.4 -0.4 1 0 1 0 0.4 -0.4 CHG methylation CG methylation CAC1 DNA/En-Spm F1 CG hypo DMR density 0 1 5’ 3’ Col Cxm mxC F2 Cxm F2 mxC met1 1 2 1 2 1 2 3 1 2 3 CAC1 presence absenceF
C
B
D
Col met1 Cxm - rF1 mxC - rF1Fig. 5. Hypomethylation at TEs in epihybrids. (A) Average distribution of CG, CHG, and CHH methyl-ation in indicated genotypes over VANDAL elements containing at least one F1 CHH and CHG hypo-DMR. (B) Distribution of CG hypomethylation DMR density over DNA/En-Spm elements in F1 epihybrids. (C) Ge-nome-browser view of CG and CHG methylation at CAC1. Col and met1 parental methylation patterns are shown together with the rF1 profile subtracted from the C×m or the m×C F1 profiles. The dashed boxes show regions with CG and CHG hypomethylation in epihybrids. (D) PCR assay of CAC1 excision in indicated genotypes. Amplification of the“absence” band reflects mobilization of the TE. (E) CHG methyl-ation levels at CHG hypomethylmethyl-ation DMRs that show transgressive behavior in F1 epihybrids. Values were extracted from one replicate per genotype. (F) CHG hypomethylation DMR density over LTR/Gypsy elements in F1 epihybrids. PLANT BIOLO GY PNAS PLUS
epihybrids correlated with mobilization of this element in the F2 progeny (Fig. 5D). Together, these data show that DNA hypo-methylation induced in F1 epihybrids, potentially involving the TE-encoded TnpA protein, can result in mobilization of ele-ments that did not transpose in either parent.
Transgressive Hypomethylation of LTR/Gypsy Elements in F1 Epihybrids.
Transgressive CHG hypomethylation was specifically initiated in F1 epihybrids at one thousand genomic regions, which were highly methylated in bothMET1+andmet1 parents (Fig. 5E andSI Ap-pendix, Fig. S11A) and were overrepresented for long LTR/Gypsy elements (mean length of 6.4 kb) (SI Appendix, Fig. S11 B and C). Transgressive CHG hypomethylation preferentially occurred in internal coding regions of LTR/Gypsy TEs, with a bias toward the 3′ half of the elements (Fig. 5F), and it was accompanied by decreased CHH methylation within TE bodies, with little change at TE edges (SI Appendix, Fig. S11D). CMT2 targets the body of long TEs for CHH methylation, and to a lesser extent for CHG methylation (9, 10).CMT2 transcripts were reduced threefold inmet1, potentially contributing to decreased non-CG methylation at long TEs whereas CMT2 transcripts ac-cumulated at a midparental level in F1 plants (SI Appendix, Fig. S11E). Reduced CMT2 expression relative to MET1+ plants may be at the origin of CHH hypomethylation at in-ternal sequences of long TEs in F1 epihybrids. Collectively, our results reveal widespread DNA hypomethylation at both DNA transposons and retroelements in F1 epihybrids.
Transcription Dynamics in F1 Epihybrids. It is generally accepted that DNA methylation can influence RNA expression although the mechanisms are often unclear. Because of the profound modification of DNA methylation patterns occurring in F1 epi-hybrids, we determined how DNA methylation correlated with genome-wide mRNA profiles of F1 epihybrids and their parents. At the chromosomal level, the strongly increased transcript lev-els in pericentromeric regions were largely unchanged in F1 epihybrids (SI Appendix, Fig. S12A), extending previous reports of inheritance of met1-induced release of silencing at selected loci (7, 15, 34). Additional changes in transcript profiles in-dicated, however, that the silencing/desilencing states of many loci are not necessarily stably inherited by the F1. Overall gene tran-script levels varied only modestly between parents and F1 epihy-brids, independent of DNA methylation changes in F1 (Fig. 6A andSI Appendix, S12B). In contrast, TEs that were strongly acti-vated in met1 were expressed at levels above the MPV in F1 epihybrids, and both TEs overlapping with F1 CG and CHG hypo-DMRs and F1 CHH hyper-hypo-DMRs showed increased expression relative to the MPV (Fig. 6A andSI Appendix, S12B). These data indicate that changes in DNA methylation in epihybrids are mostly associated with further derepression of TE silencing.
Combining two complementary approaches to determine dif-ferential expression between samples, we identified 3,606 up-regulated and 2,169 down-up-regulated loci inmet1 compared with theMET1+parent (SI Appendix, Fig. S12C), substantially adding to the number of met1-misexpressed loci previously identified (4–6, 39). The large majority (more than 85%) of loci mis-regulated inmet1 behaved in a similar manner in both F1 epi-hybrids (Fig. 6B). Of the 1,576 protein-coding gene transcripts up-regulated inmet1, about 40% accumulated at an intermediate level in epihybrids, indicating stable inheritance ofmet1-induced transcriptional derepression in epihybrids. This set of genes in-cludes, and behaves similarly to, the archetypeFWA gene, which remains transcriptionally derepressed and hypomethylated upon restoration of the MET1 function (7). Expression of another 40% ofmet1-derepressed genes was corrected in epihybrids (Fig. 6B), revealing efficient reestablishment of transcriptional repression. The remaining genes, about 240, continued to be up-regulated in F1 epihybrids atmet1-like levels. Independently of their behavior in F1 epihybrids, genes up-regulated in met1 were rarely reme-thylated in F1 epihybrids (SI Appendix, Fig. S13A), indicating that
pend on DNA methylation. There were two notable exceptions: SDC, a well-known target of DNA methylation-dependent si-lencing (40), andKELP (AT4G10920), which encodes a target of tobamoviruses (41), were efficiently resilenced in F1 epihybrids. Resilencing correlated with DNA remethylation of tandem re-peats located in the promoter regions of both genes (SI Appendix, Fig. S14).
Similar to what we had observed forIBM1 and ROS1, expression of mostmet1–down-regulated genes was not restored to MET1+ levels in F1 epihybrids (Fig. 6B). Thus, similar to transcriptional activation,met1-induced transcriptional repression can be inheri-ted. Transcriptional down-regulation in met1 was not associated with hypermethylation, and the F1 epihybrid methylation pattern at these genes was similar to the expected rF1, independent of their F1 epihybrid expression level (SI Appendix, Fig. S13B). These results indicate that transcriptional changes at these genes in F1 epihybrids mostly result from indirect effects rather than alter-ations of their DNA methylation patterns.
That resilencing of about 40% of genes up-regulated inmet1 is independent of remethylation in F1 epihybrids indicates that the mechanism preventing formation of stable epialleles at these genes does not rely on DNA methylation. For the remaining genes that are up-regulated inmet1, and for most met1-repressed genes, the met1 transcriptional state is stably inherited or exac-erbated in F1 epihybrids. Reexamining published data (42), we found that only genes that were up-regulated inmet1 but that were expressed at an intermediate or met1-like level in F1 epi-hybrids were enriched in H2A.Z inmet1 (SI Appendix, Fig. S15), even though all genes equally lost CG methylation in met1 (SI Appendix, Fig. S13). This observation suggests that H2A.Z enrichment caused by CG methylation depletion (42, 43) possi-bly plays a role in stabilizing transcriptional derepression upon restoration of MET1 function in epihybrid plants. Additionally, because H2A.Z enrichment correlates with responsiveness of gene expression to environmental or developmental factors (43), these transcriptionally activated epialleles may acquire new sensitivity to such stimuli.
Further Release of TE Silencing in F1 Epihybrids.In sharp contrast with genes, very few TEs derepressed inmet1 were resilenced in F1 epihybrids: only 55 out of 1,921 (Fig. 6B). Unexpectedly, more than half ofmet1-activated TEs accumulated to met1-like or even higher levels in F1 epihybrids whereas only a third were expressed at MPV, reflecting stable inheritance of their altered transcriptional state in F1 epihybrids (Fig. 6B). For instance, the MET1+-derived, hypomethylated copies ofHi and CAC1 (Fig. 5C and SI Appendix, Fig. S10A) were expressed at met1-like levels in F1 epihybrids (SI Appendix, Fig. S16A), suggesting re-lease of silencing at the correspondingMET1+-derived alleles in the F1 epihybrids.
Overall, TEs hyperactivated in F1 epihybrids were enriched for LTR retroelements, in particular pericentromeric LTR/Gypsy ATHILA retroelements (SI Appendix, Fig. S16B), which showed CHH hypomethylation in their body and strong transgressive CHG hypomethylation (Fig. 6C and D). Transcript accumulation in F1 epihybrids was especially high for the 3′ half of LTR/Gypsy elements (SI Appendix, Fig. S16C), a region known as Tran-scriptionally Silent Information (TSI) at some ATHILA ele-ments (44), which also displayed preferential transgressive CHG hypomethylation in F1 epihybrids (Fig. 5F). Besides LTR/ Gypsy retroelements, several LTR/Copia retrotransposons, in-cluding ATCOPIA48 (AT1TE66380), were transgressively up-regulated in F1 epihybrids (Fig. 6E and SI Appendix, S16B). ATCOPIA48 LTRs are rich in CG and CHG sites, with average MET1+methylation levels of 82% and 46% at the 11 CG and 17 CHG sites (Fig. 6F). ATCOPIA48 was expressed at a very low level inmet1 despite loss of CG methylation, indicating that release of silencing in F1 epihybrids likely reflects alteration in an additional repressive mark. CHG methylation was reduced at theATCOPIA48 5′ LTR in F1 epihybrids (Fig. 6F). Using published data (45), we
found thatATCOPIA48 is strongly activated in cmt3 and suvh4/5/6 mutants (Fig. 6E), revealing that its silencing state depends on CHG methylation. Therefore, derepression ofATCOPIA48 in F1 epihybrids likely results from transgressive CHG hypomethylation. Collectively, our results show that silencing is further released at numerous TEs in F1 epihybrids in association with transgressive CHG hypomethylation.
Immediate Resilencing at TEs in F1 Epihybrids.The few TEs with MET1+-like expression in F1 epihybrids were enriched for DMRs that were CG-hypermethylated relative to the rF1 (Fig. 6D), sug-gesting that resilencing at these elements relies on CG remethy-lation. Silencing of the EVD and ATCOPIA21 retroelements, which are expressed and mobilized inmet1, was efficiently rees-tablished in F1 epihybrids (Fig. 6G andSI Appendix, Fig. S10C) although theEVD 5′ LTR, which is almost exclusively methylated at CG sites (19), was not remethylated in F1 epihybrids (Fig. 6H). ATCOPIA21 LTRs, which lack CG and CHG sites, have around 25% CHH methylation inMET1+, which is nearly lost in met1 mutants. Similar toEVD, ATCOPIA21 5′ LTRs were not remethy-lated in either reciprocal F1 epihybrid (Fig. 6H). Thus, these results reveal that resilencing of active autonomous TEs originally repressed by DNA methylation, and of their neocopies, can occur rapidly and independently of DNA remethylation. Future work needs to de-termine the molecular mechanism by which this resilencing occurs.
Changes in 24-nt sRNA Populations and DNA Methylation Changes in F1 Epihybrids.In plants, small RNAs (sRNAs) are involved in the establishment of DNA methylation and part of its maintenance
in asymmetric context. We analyzed small RNA populations, and monitored the activity of loci with differential 24-nt sRNA ac-cumulation between parents in the F1 epihybrids. Inmet1, there were about twice as many regions of 24-nt sRNA over-accumu-lation (13,871) as of regions of decreased 24-nt sRNA abun-dance (6,582) (SI Appendix, Fig. S17A), with the magnitude of loss, on average 99-fold, being much more severe than the magnitude of gain, 13-fold, accounting for the reduction in 24-nt sRNAs in met1 (SI Appendix, Fig. S17B). The levels of 24-nt sRNAs deviated at a few regions from the MPV; regions with reduced 24-nt sRNA accumulation were enriched for MuDR TEs, and regions with increased accumulation for LTR/Gypsy TEs (SI Appendix, Fig. S17 A and C).
DNA hypermethylation in F1 epihybrids was associated with increased, often transgressive 24-nt sRNA levels in epihybrids (SI Appendix, Fig. S18A). Conversely, DMRs that were hypo-methylated in F1 epihybrids accumulated few 24-nt sRNAs in both met1 and in F1 epihybrids, suggesting that 24-nt sRNA production becomes impaired at the corresponding MET1+ -derived alleles in F1 epihybrids (SI Appendix, Fig. S18B). For instance, VANDAL elements that are hypomethylated in F1 epihybrids featured drastically reduced levels of 24-nt sRNA in epihybrid plants (SI Appendix, Fig. S18D). These results show that DNA methylation changes at TEs in F1 epihybrids are often associated with changes in 24-nt local sRNA accumulation.
In contrast to TE hypermethylation, CHG hypermethylation at genes in F1 epihybrids, including IBM1 targets BNS and AT3G20790, was not associated with accumulation of sRNAs, either in the parents or in F1 epihybrids (SI Appendix, Fig. S19). −4 0 4 log2 FC test/MPV hypo CG hypo CHG hypo CHH hyper CG hyper CHG hyper CHH F1 DMRs genes Cxm mxC
A
# lociB
0 400 800 1200 Up met1-like Inter. WT-like TE down in met1 F1 transcript level gene up in met1 TE up in met1 gene down in met1−4 0 4 log2 FC test/MPV TEs hypo CG hypo CHG hypo CHH hyper CG hyperCHG hyper CHH F1 DMRs Cxm mxC 15 55 4 18 ) %( noi tal yht e m G H C ) %( noi tal yht e m H H C -2kb +2kb TE -2kb +2kb TE Colmet1rF1 CxmmxC
C
100 0 ATCOPIA48 Col met1 Cxm mxC cmt3 suvh4/5/6 mRNAE
0 80 CG CHG CHH Col met1 rF1 Cxm mxC ) %( noi tal yht e m ATCOPIA48 5’ LTRF
30 0 Col met1 Cxm mxC 20 0 EVD ATCOPIA21 mRNAG
0 60 CG methylation (%) EVD 5’LTR 0 25 CHH methylation (%) ATCOPIA21 5’LTR Col met1 rF1 Cxm mxCH
0 1.6resilenced Inter. met1-like Up hypo-CHH hyper-CHG hyper-CHH hypo-CG hyper-CG hypo-CHG average # of DMRs per TE
D
F1 transcript levelFig. 6. RNA expression in F1 epihybrids. (A) RNA expression at loci overlapping with different DMR types in F1 epihybrids relative to MPV. (B) Mis-regulated genes and TEs in met1 grouped according to their transcription level in epihybrids. Inter., in-termediate. Only genes with similar expression in both reciprocal F1 epihybrids are shown. (C) Average distribution of CHG and CHH methylation over TEs activated in met1 and transcribed at a met1-like or higher level in F1 epihybrids. (D) Average number of different DMRs overlapping with TEs grouped according to their transcript level in epihybrids. Inter., intermedi-ate. (E) Genome browser view of mRNA accumu-lation at ATCOPIA48 (AT1TE66380) in indicated genotypes. RNA levels in cmt3 and suvh4/5/6 are from ref. 45. (F ) DNA methylation levels at the 5′ LTR of ATCOPIA48. (G) Genome-browser view of mRNA accumulation at EVD (AT5TE20395) and ATCOPIA21 (AT5TE65370) elements in indicated genotypes. (H) DNA methylation levels at the 5′ LTRs of EVD and ATCOPIA21 elements. PLANT BIOLO GY PNAS PLUS
impaired IBM1 activity and/or H3K9me2 enrichment, rather than by an sRNA-mediated process, although sRNAs might be transitorily produced and required for DNA methylation during an earlier developmental window. Similarly,ddm1-induced BNS DNA hypermethylation depends on H3K9me2 and not on sRNAs (46).
Differential Inheritance ofmet1-Induced easiRNA Production in F1 Epihybrids. When transcriptionally activated, a number of TEs generate 21-nt epigenetically activated siRNAs, or easiRNAs (4, 47–49). met1 leaves, similar to flower buds (4), were character-ized by a global overaccumulation of 21-nt sRNAs matching TEs, suggesting that production of easiRNAs also takes place in the met1 sporophyte (SI Appendix, Figs. S17B and S20A). Regions of increased abundance of 21-nt sRNAs in met1 leaves mostly overlapped with TEs from the LTR/Gypsy and En-Spm super-families (SI Appendix, Fig. S20B). Only a few 21-nt sRNA loci that deviated significantly from the MPV were identified in F1 epihybrids (SI Appendix, Fig. S20A), suggesting that easiRNA production frommet1-derived TE alleles is generally inherited in F1 plants. However, out of the 42 En-Spm elements producing easiRNA in met1, easiRNA accumulation in reciprocal F1 epi-hybrids was reduced at 32 elements, half of which beingATENSPM5 elements (SI Appendix, Fig. S20 A and C).
Similar to ddm1 inflorescences (50), the ATHILA6A and ATHILA2 LTR-retrotransposon families made up the bulk of easiRNAs inmet1 leaves whereas ATENSPM5 was the top DNA TE family (SI Appendix, Fig. S20C). Accumulation of easiRNAs was biased toward the 3′ TSI part of ATHILA6A elements and toward the transposase-encoding ORF1 ofATENSPM5 elements (Fig. 7A). Production of easiRNAs from these TEs was, however, radically different in F1 epihybrids. Although the overall ATHILA6A easiRNA levels were near the MPV, ATENSPM5 elements accumulated virtually no easiRNAs in F1 epihybrids (Fig. 7A). The loss of easiRNA was associated with efficient resilencing of theATENSPM5 ORF1 in F1 epihybrids whereas the second ORF, which produced no or only very few easiRNA inmet1, remained transcriptionally active (Fig. 7 A and B). At ATHILA6A elements, the 3′ TSI region was strongly derepressed inmet1 and remained transcribed at an MPV in F1 epihybrids (Fig. 7B). Resilencing of ATENSPM5 ORF1 in F1 epihybrids correlated with DNA hypermethylation in all cytosine con-texts and with increased 24-nt sRNA accumulation (Fig. 7C and SI Appendix, Fig. S20D). ATHILA6A elements, which accumulated high levels of 24-nt sRNA in met1 and F1 epihy-brids, were instead hypomethylated at CHG sites in epihybrids (Fig. 7C andSI Appendix, Fig. S20D). EasiRNAs have previously been implicated in reestablishment of silencing at TEs after ddm1-induced transcriptional activation and restoration of func-tional DDM1 protein by outcrossing (50). Together, our results demonstrate a complex relationship between easiRNA and/or 24-nt sRNAs produced from differe24-nt TEs activated inmet1, DNA methylation, and resilencing in F1 epihybrids. At the
trans-posase-encoding ORF1 of ATENSPM5, easiRNAs can likely
efficiently initiate DNA hypermethylation early during devel-opment of F1 epihybrids. Upon reestablishment of silencing, DNA hypermethylation leads to high 24-nt sRNA production, which then takes over to ensure the maintenance of silencing throughout development. We note thatATHILA elements, which are not resilenced in F1 epihybrids, are nonautonomous retro-transposons whereas most easiRNA-producing ATENSPM5 ele-ments are long intact DNA TEs.
Discussion
When normally methylated WT chromosomes and extensively demethylated met1 chromosomes meet in F1 epihybrids, an epigenomic shock is triggered that is characterized by trans-gressive DNA methylation and gene expression. Most of the deviation from MPV in F1 epihybrids is independent of the di-rection of the cross. The epigenomic shock results in the creation
have reduced or increased non-CG methylation and expression. Particularly notable is the formation of a new nonparental hypomethylated, stable IBM1 epiallele with low expression. A quarter of the previously describedmet1-epiRIL lines, which are MET1+ progeny of self-pollinated F1 epihybrids (34), should be homozygous for this IBM1 epiallele. Because IBM1 protects several thousand genes from H3K9me2 and associated CHG methylation, the nonparentalIBM1 epiallele likely contributes to the appearance of nonparental genome-wide methylation pat-terns that have been observed in these lines.
Most of the deviation from MPV in F1 epihybrids occurs at TEs. Many pericentromeric elements, including many Helitrons (SI Appendix, Fig. S8A), experience immediate CG remethyla-tion in F1 epihybrids. At these elements, DNA methylaremethyla-tion, in-cluding CG methylation, is likely tightly dependent on sRNAs because these loci are associated with high levels of 24-nt sRNAs and retain a certain level of CG methylation even in themet1-3 null mutant. However, CG remethylation is not observed at all loci associated with high levels of 24-nt sRNAs, suggesting that remethylated loci must have specific properties. Immediate CG remethylation in epihybrids, potentially involving sRNA-directed MET1-dependent de novo methylation (51), may be stimulated by remaining CG methylation at themet1-derived alleles and/or by specific sequence motifs or chromatin features. Either way, immediate CG remethylation in epihybrids prevents persistence of stable hypomethylated epialleles at these loci. Previous anal-yses of met1-derived epiRILs in the F9 generation have sug-gested active remethylation of centromeric DNA at CG and/or CHG sites (34). Our results support this conclusion and further reveal that pericentromeric regions are efficiently targeted for rapid CG remethylation as soon as MET1 activity is restored in the F1 epihybrids. In contrast, DNA remethylation of TEs lo-cated throughout the genome occurred only over several gener-ations in ddm1-epiRILs (36), indicating that remethylation after outcrossing ofmet1 or ddm1 mutations involves at least partly dis-tinct mechanisms. That parental CG methylation patterns within gene bodies are overall faithfully inherited in F1 epihybrids (SI Appendix, Fig. S3B) suggests the absence of a corrective mechanism that can reestablish gene-body CG methylation. It is possible that remethylation eventually occurs, but only after multiple generations.
Despite DNA remethylation, only fewmet1-activated TEs are resilenced in F1 epihybrids, and the two LTR retrotransposons EVD and ATCOPIA21, which are active in the met1 parent, are transcriptionally resilenced in F1 epihybrids independently of DNA remethylation of their LTRs. Therefore, the epigenomic shock also triggers the activity of an enigmatic silencing mech-anism that was not active in the parents. Whether EVD and ATCOPIA21 acquire the same silencing state in F1 epihybrids and what the molecular nature of this state is remain to be de-termined. Restoration of silencing atEVD in epihybrids is sur-prising, given that EVD remains mobile in late generations of somemet1 epiRIL lines (19), suggesting that the silenced state established in F1 epihybrids can be easily reversed.
In F1 epihybrids, DNA hypomethylation at numerous TEs is associated with enhanced release of silencing or transcriptional
activation. At some, including VANDAL and CACTA
super-family members, hypomethylation in F1 epihybrids likely initiates through transdemethylation of MET1+-derived TEs, mediated by TE-encoded factors produced from the active met1-inherited elements. We also detected transgressive CHG hypomethylation at numerous TEs in F1 epihybrids, especially at many LTR/ GypsyATHILA elements, which are highly CHG-methylated in bothMET1+andmet1 parents. It has been previously shown that only when TEs are transcriptionally activated can they become targets of IBM1-mediated demethylation of H3K9me2, resulting in CHG hypomethylation and further transcriptional activation (52). Accordingly, forced IBM1 expression in met1 leads to transcriptional overactivation of some TEs (23). IBM1 expres-sion is compromised inmet1, and, despite high H3K9me2 and
CHG methylation levels, these TEs are active. We propose that, in F1 epihybrids, active transcription of these TEs may preferentially recruit the limited amount of IBM1 available from theMET1+-derived IBM1 allele, resulting in H3K9me2 and CHG demethylation and thus further transcriptional de-repression. Supporting this view, TEs expressed at or above met1-levels in F1 epihybrids, which are overrepresented forATHILA elements, had a higher-than-average density of CHG sites (SI Appendix, Fig. S16D), a feature known to affect sensitivity toibm1 mutations (52). Long TEs are also CHH-hypomethylated in F1 epihybrids, especially in their center, which may be caused by reducedCMT2 activity. Because CMT2 mediates CHH methyl-ation through binding to H3K9me2 (9), CMT2 recruitment to these TEs may be impaired due to the initial reduction in H3K9me2 and CHG methylation. Additionally, because CHH methylation is strongly required for H3K9me2 at CMT2-target sites (9), defective CMT2 targeting may contribute to the transgressive reduction in CHG methylation at long TEs in F1 epihybrids. Our data support a scenario in which transcriptional up-regulation ofATHILA TEs with transgressive CHG hypo-methylation in epihybrids would occur at already transcriptionally active TE copies inherited from the met1 parent.
Transcrip-tional activation of ATHILA elements has been observed
in interspecificArabidopsis thaliana × arenosa hybrids (53). Supporting our hypothesis, onlyATHILA elements contributed by theArabidopsis arenosa parent, which already show a low level of expression in this species, are up-regulated in these hybrids (53).
Intraspecific F1 hybrids derived from crosses between the A. thaliana accessions C24 and Ler deviate from MPV DNA methylation at numerous loci, along with a global decrease in 24-nt sRNA accumulation (54, 55). In these hybrids, mostly genic and gene-proximal regions are affected, in sharp contrast with the situation in our F1 epihybrids, in which the majority of DNA methylation effects are seen at TEs. Transgressive DNA methylation was, however, not found in C24× Ler F1 hybrids, suggesting that the epigenomic shock is triggered only when parental chromosomes differ markedly in DNA methylation level, and especially at TEs. TE methylation is likely similar between C24 and Ler because previous studies have pointed out that methyl-ation of TE sequences is highly stable, both across genermethyl-ations and betweenA. thaliana accessions (56–59).
It has been suggested that both genetic and epigenetic dif-ferences between parents contribute to hybrid vigor, or heterosis. Although met1 and MET1+ plants differ drastically in DNA methylation, sRNA accumulation, and gene expression, the F1 epihybrids are not obviously superior in biomass or rosette di-ameter (SI Appendix, Fig. S21). We have not yet measured other traits, such as pathogen or stress tolerance, but this observation already calls into question simplistic models for the contribution of epigenetic differences to heterosis.
Various stresses or environmental conditions can modulate patterns of DNA methylation and/or activate TE transcription (39, 60). Further, mutations altering the function of DNA methylation regulators may spontaneously arise in natural pop-ulations, generating individuals with partially hypomethylated genomes and even activated TEs (61–63). Although A. thaliana is a mainly self-fertilizing species, its effective outcrossing rate varies considerably between different stands and can be higher than 10% (64). Thus, it is plausible that epigenomic shocks, al-though likely less drastic than the one we report here, may occur in the wild and generate novel heritable epigenetic diversity and associated regulatory patterns. Given their impact on TEs, such shocks may also have important implications for genome-wide TE dynamics.
Materials and Methods
Second generation met1-3 mutant plants were used in this study (8). All experiments were performed starting from 3-wk-old leaf material. An extended description of materials and methods, including poly(A)-RNA sequencing, whole-genome bisulfite sequencing, small RNA sequencing, RT-PCR analysis, and locus-specific DNA methylation analysis is provided in
SI Appendix, SI Materials and Methods. Sequencing data generated in this study have been deposited with the European Nucleotide Archive (ENA,
www.ebi.ac.uk/ena) under accession number PRJEB9919.
ACKNOWLEDGMENTS. This work was supported by the Max Planck Society and Deutsche Forschungsgemeinschaft SFB 1101 (to D.W.), the Centre Na-tional de la Recherche Scientifique (O.M.), the European Community’s Sev-enth Framework Programme through a Starting Independent Research Grant from the European Research Council (I2ST, 260742) (to O.M.), and a Young Researcher Grant from the Auvergne Regional Council (to O.M.). O.M. is a young investigator of the European Molecular Biology Organiza-tion (EMBO). 0 25 0 25 5 0 5 -1kb +1kb ATHILA6A -1kb +1kb -1kb +1kb ATENSPM5 -1kb +1kb Col met1 Cxm mxC 0 80 ORF1 ORF2 RPM normalized 21-nt sRNA count 0 500 RPM 0 100 0 60 0 18 ATENSPM5 CG methylation (%) CHG methylation (%) CHH methylation (%) 0 90 5 20 0 65 ATHILA6A normalized 21-nt sRNA count b k 1 + b k 1 + b k 1 - -1kb Col met1 Cxm mxC Colmet1rF1CxmmxC Colmet1rF1CxmmxC
A
B
C
5 M P S N E T A A 6 A L I H T A 2 F R O 1 F R O 2 F R O 1 F R O 2 F R O 1 F R O 2 F R O 1 F R OFig. 7. The 21-nt sRNA accumulation, mRNA, and DNA methylation levels at ATHILA6A and ATENSPM5 elements. (A) Accumulation of 21-nt sRNAs along ATHILA6A and ATENSPM5 elements in parent (Top) and F1 epihybrids (Bottom). The two ORFs in both TEs are depicted as yellow arrows. (B) Box plots of mRNA accumulation at ATHILA6A (Left) and the two ATENSPM5 ORFs (Right) in indicated genotypes. (C) Average distribution of CG, CHG, and CHH methylation in indicated genotypes over ATHILA6A and ATENSPM5 elements producing 21-nt sRNAs in met1.
PLANT
BIOLO
GY
PNAS
patterns in plants and animals. Nat Rev Genet 11(3):204–220.
2. Rigal M, Mathieu O (2011) A“mille-feuille” of silencing: Epigenetic control of transposable elements. Biochim Biophys Acta 1809(8):452–458.
3. Cokus SJ, et al. (2008) Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452(7184):215–219.
4. Lister R, et al. (2008) Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133(3):523–536.
5. Zhang X, et al. (2006) Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell 126(6):1189–1201.
6. Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S (2007) Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence be-tween methylation and transcription. Nat Genet 39(1):61–69.
7. Kankel MW, et al. (2003) Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 163(3):1109–1122.
8. Saze H, Mittelsten Scheid O, Paszkowski J (2003) Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat Genet 34(1): 65–69.
9. Stroud H, et al. (2014) Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat Struct Mol Biol 21(1):64–72.
10. Zemach A, et al. (2013) The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 153(1):193–205. 11. Du J, et al. (2014) Mechanism of DNA methylation-directed histone methylation by
KRYPTONITE. Mol Cell 55(3):495–504.
12. Du J, et al. (2012) Dual binding of chromomethylase domains to H3K9me2-containing nucleosomes directs DNA methylation in plants. Cell 151(1):167–180.
13. Vongs A, Kakutani T, Martienssen RA, Richards EJ (1993) Arabidopsis thaliana DNA methylation mutants. Science 260(5116):1926–1928.
14. Miura A, et al. (2001) Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature 411(6834):212–214.
15. Lippman Z, May B, Yordan C, Singer T, Martienssen R (2003) Distinct mechanisms determine transposon inheritance and methylation via small interfering RNA and histone modification. PLoS Biol 1(3):E67.
16. Tsukahara S, et al. (2009) Bursts of retrotransposition reproduced in Arabidopsis. Nature 461(7262):423–426.
17. Singer T, Yordan C, Martienssen RA (2001) Robertson’s Mutator transposons in A. thaliana are regulated by the chromatin-remodeling gene Decrease in DNA Meth-ylation (DDM1). Genes Dev 15(5):591–602.
18. Fu Y, et al. (2013) Mobilization of a plant transposon by expression of the transposon-encoded anti-silencing factor. EMBO J 32(17):2407–2417.
19. Mirouze M, et al. (2009) Selective epigenetic control of retrotransposition in Arabidopsis. Nature 461(7262):427–430.
20. Kato M, Miura A, Bender J, Jacobsen SE, Kakutani T (2003) Role of CG and non-CG methylation in immobilization of transposons in Arabidopsis. Curr Biol 13(5):421–426. 21. Stroud H, Greenberg MV, Feng S, Bernatavichute YV, Jacobsen SE (2013) Compre-hensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152(1-2):352–364.
22. Mathieu O, Reinders J, Caikovski M, Smathajitt C, Paszkowski J (2007) Transgenera-tional stability of the Arabidopsis epigenome is coordinated by CG methylation. Cell 130(5):851–862.
23. Rigal M, Kevei Z, Pélissier T, Mathieu O (2012) DNA methylation in an intron of the IBM1 histone demethylase gene stabilizes chromatin modification patterns. EMBO J 31(13):2981–2993.
24. Saze H, Shiraishi A, Miura A, Kakutani T (2008) Control of genic DNA methylation by a jmjC domain-containing protein in Arabidopsis thaliana. Science 319(5862): 462–465.
25. Saze H, et al. (2013) Mechanism for full-length RNA processing of Arabidopsis genes containing intragenic heterochromatin. Nat Commun 4:2301.
26. Deleris A, et al. (2012) Loss of the DNA methyltransferase MET1 Induces H3K9 hy-permethylation at PcG target genes and redistribution of H3K27 trimethylation to transposons in Arabidopsis thaliana. PLoS Genet 8(11):e1003062.
27. Mathieu O, Probst AV, Paszkowski J (2005) Distinct regulation of histone H3 meth-ylation at lysines 27 and 9 by CpG methmeth-ylation in Arabidopsis. EMBO J 24(15): 2783–2791.
28. Stokes TL, Kunkel BN, Richards EJ (2002) Epigenetic variation in Arabidopsis disease resistance. Genes Dev 16(2):171–182.
29. Saze H, Kakutani T (2007) Heritable epigenetic mutation of a transposon-flanked Arabidopsis gene due to lack of the chromatin-remodeling factor DDM1. EMBO J 26(15):3641–3652.
30. Jacobsen SE, Meyerowitz EM (1997) Hypermethylated SUPERMAN epigenetic alleles in arabidopsis. Science 277(5329):1100–1103.
31. Soppe WJ, et al. (2000) The late flowering phenotype of fwa mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Mol Cell 6(4):791–802.
stable inheritance of DNA hypomethylation induced by ddm1 mutation of Arabi-dopsis thaliana. Genetics 151(2):831–838.
33. Jacobsen SE, Sakai H, Finnegan EJ, Cao X, Meyerowitz EM (2000) Ectopic hyper-methylation of flower-specific genes in Arabidopsis. Curr Biol 10(4):179–186. 34. Reinders J, et al. (2009) Compromised stability of DNA methylation and transposon
immobilization in mosaic Arabidopsis epigenomes. Genes Dev 23(8):939–950. 35. Johannes F, et al. (2009) Assessing the impact of transgenerational epigenetic
vari-ation on complex traits. PLoS Genet 5(6):e1000530.
36. Teixeira FK, et al. (2009) A role for RNAi in the selective correction of DNA methyl-ation defects. Science 323(5921):1600–1604.
37. Cui H, Fedoroff NV (2002) Inducible DNA demethylation mediated by the maize Suppressor-mutator transposon-encoded TnpA protein. Plant Cell 14(11):2883–2899. 38. Schläppi M, Raina R, Fedoroff N (1994) Epigenetic regulation of the maize Spm transposable element: Novel activation of a methylated promoter by TnpA. Cell 77(3): 427–437.
39. Dowen RH, et al. (2012) Widespread dynamic DNA methylation in response to biotic stress. Proc Natl Acad Sci USA 109(32):E2183–E2191.
40. Henderson IR, Jacobsen SE (2008) Tandem repeats upstream of the Arabidopsis en-dogene SDC recruit non-CG DNA methylation and initiate siRNA spreading. Genes Dev 22(12):1597–1606.
41. Matsushita Y, Deguchi M, Youda M, Nishiguchi M, Nyunoya H (2001) The tomato mosaic tobamovirus movement protein interacts with a putative transcriptional co-activator KELP. Mol Cells 12(1):57–66.
42. Zilberman D, Coleman-Derr D, Ballinger T, Henikoff S (2008) Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature 456(7218):125–129. 43. Coleman-Derr D, Zilberman D (2012) Deposition of histone variant H2A.Z within gene
bodies regulates responsive genes. PLoS Genet 8(10):e1002988.
44. Steimer A, et al. (2000) Endogenous targets of transcriptional gene silencing in Arabidopsis. Plant Cell 12(7):1165–1178.
45. Stroud H, et al. (2012) DNA methyltransferases are required to induce heterochro-matic re-replication in Arabidopsis. PLoS Genet 8(7):e1002808.
46. Sasaki T, Kobayashi A, Saze H, Kakutani T (2012) RNAi-independent de novo DNA methylation revealed in Arabidopsis mutants of chromatin remodeling gene DDM1. Plant J 70(5):750–758.
47. Slotkin RK, et al. (2009) Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 136(3):461–472.
48. Tanurdzic M, et al. (2008) Epigenomic consequences of immortalized plant cell sus-pension culture. PLoS Biol 6(12):2880–2895.
49. Creasey KM, et al. (2014) miRNAs trigger widespread epigenetically activated siRNAs from transposons in Arabidopsis. Nature 508(7496):411–415.
50. Nuthikattu S, et al. (2013) The initiation of epigenetic silencing of active transposable elements is triggered by RDR6 and 21-22 nucleotide small interfering RNAs. Plant Physiol 162(1):116–131.
51. Aufsatz W, Mette MF, Matzke AJ, Matzke M (2004) The role of MET1 in RNA-directed de novo and maintenance methylation of CG dinucleotides. Plant Mol Biol 54(6):793–804. 52. Inagaki S, et al. (2010) Autocatalytic differentiation of epigenetic modifications
within the Arabidopsis genome. EMBO J 29(20):3496–3506.
53. Josefsson C, Dilkes B, Comai L (2006) Parent-dependent loss of gene silencing during interspecies hybridization. Curr Biol 16(13):1322–1328.
54. Groszmann M, et al. (2011) Changes in 24-nt siRNA levels in Arabidopsis hybrids suggest an epigenetic contribution to hybrid vigor. Proc Natl Acad Sci USA 108(6): 2617–2622.
55. Greaves IK, et al. (2012) Trans chromosomal methylation in Arabidopsis hybrids. Proc Natl Acad Sci USA 109(9):3570–3575.
56. Becker C, et al. (2011) Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 480(7376):245–249.
57. Schmitz RJ, et al. (2011) Transgenerational epigenetic instability is a source of novel methylation variants. Science 334(6054):369–373.
58. Schmitz RJ, et al. (2013) Patterns of population epigenomic diversity. Nature 495(7440):193–198.
59. Vaughn MW, et al. (2007) Epigenetic natural variation in Arabidopsis thaliana. PLoS Biol 5(7):e174.
60. Tittel-Elmer M, et al. (2010) Stress-induced activation of heterochromatic transcrip-tion. PLoS Genet 6(10):e1001175.
61. Dubin MJ, et al. (2015) DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. eLife 4:e05255.
62. Shen X, et al. (2014) Natural CMT2 variation is associated with genome-wide meth-ylation changes and temperature seasonality. PLoS Genet 10(12):e1004842. 63. Woo HR, Pontes O, Pikaard CS, Richards EJ (2007) VIM1, a methylcytosine-binding
protein required for centromeric heterochromatinization. Genes Dev 21(3):267–277. 64. Bomblies K, et al. (2010) Local-scale patterns of genetic variability, outcrossing, and spatial structure in natural stands of Arabidopsis thaliana. PLoS Genet 6(3):e1000890.