HAL Id: hal-02846226
https://hal.archives-ouvertes.fr/hal-02846226
Submitted on 25 Jun 2020
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
NMR and ESR study of the conformations and
dynamical properties of poly(L-lysine) in aqueous
solutions
B. Perly, Yves Chevalier, C. Chachaty
To cite this version:
B. Perly, Yves Chevalier, C. Chachaty. NMR and ESR study of the conformations and dynamical
properties of poly(L-lysine) in aqueous solutions. Macromolecules, American Chemical Society, 1981,
14 (4), pp.969-975. �10.1021/ma50005a015�. �hal-02846226�
valuesofu¡i+l andZy+1were keptat3.8Á, and thoseforcZy+2,
dy+3,and ay+4were adjustedbythe triangle inequality. In
general, the valuesofuyandZyforsmall|Z
-j|havelittleeffect
in constrainingtheconformationof thewholeprotein.
(40) A cutoff distance of 10 A for both the “contact” and
“noncontact”distancesmeans thatu¡¡= 10AandL = 5Aif
d*¡j< 10A, and uy= 40Aand
Zy= 10Aif d*y> 10A. The
triangle inequalities(6)are then appliedto every setofthree
pointstomodify all ofthe u¡¡s andl¡¡s,exceptuy+1andZy+1,
whichare thenused to calculate H.
(41) Sternberg, M.J.E.;Thornton,J.M. J.Mol.Biol. 1976,105, 367. Ibid.1977,110, 269, 285.
(42) Richardson, J.S.Nature(London) 1977,268, 495.
(43) Chothia,C.;Levitt,M.;Richardson, D. Proc.Natl.Acad. Sci. U.S.A. 1977, 74, 4130.
(44) Richmond, T.J.;Richards,F.M. J.Mol.Biol. 1978,119,537. (45) Cohen,F.E.; Sternberg, M.J.E.;Taylor,W. R.Nature
(Lon-don.) 1980,285, 378.
(46) Shannon,C.E.BellSyst. Tech.J. 1948,27, 379, 623.
NMR
and
ESR
Study of the Conformations
and
Dynamical
Properties of Poly(L-lysine) in
Aqueous
Solutions
B.
Perly,*
Y.Chevalier, and
C.Chachaty
DépartementdePhysico-Chimie, Centred’Etudes Nucléaires deSaclay,
91191 Gif-sur-YvetteCedex, France. ReceivedAugust13, 1980
ABSTRACT: The conformations and dynamical behaviorofpoly(L-lysine) (PLL)inaqueoussolutionshave beeninvestigated by andl8CNMRaswellasby ESRon the end-chain spin-labeled polymer. The ESR allowedthemotionofthe macromolecular chain tobestudied up topH 13,showingthatthe random coil
-* -helix transitionat pH 11
gives rise toatwofold increaseinthecorrelationtime,with evidenceofan anisotropic reorientation. Inthe random coil state atpH7,where the segmentalmotion ofthe backbone
isquasi-isotropic, thecorrelation timegiven by ESRiscompared to thatobtained by therelaxationofthe methylprotonsofthe reduced Tempo radical residue and of thea carbons. Thedifferentmethodsyieldan activationenergyof6.5 kcalmol"1forthis motionwhereasthe frequency dependenceoftheCarelaxation
maybe interpretedbya Cole-Coledistribution of correlationtimes witha widthparameter = 0.7. The
rotationalisomerism and temperaturedependencesofinterconversion ratesofthe aminobutylsidechains
have beenanalyzed from theproton vicinalcouplings and the13Cand relaxation atdifferentfrequencies, assumingthatthe methylene groups undergo120°jumpsamongthreesites,twoofthem being equiprobable. Thesetwokindsof informationconcur toshowthatthePLLsidechainsare lessflexible thanahydrocarbon chainofsame length, possiblybecauseofthehydration ofthe NH3+terminalgroup.
Introduction
Among homopolypeptides, whichmaybeconsideredas
the simplestmodels
for natural
proteins, poly(L-lysine)(PLL)
hasbeen subjectedto a great dealof
studyon itsconformationalpropertiesas wellasitsbiological activity.1
In
aqueoussolution, poly(L-lysine) isknown to exist inseveral forms, namely,random coil,a helix, andßsheets,
depending upon
pH
andtemperature. The random coil—*·
-helix
transition whichoccurs aroundpH 11hasbeen investigatedby several
NMR
techniques,2in particularbychemicalshifts3and 13C
longitudinal
relaxation,4thelatter
method giving semiquantitative informationon the segmentalmobility of
thepolymer. More recently, poly-(L-lysine) in the-helix
formwas taken as a model in atheoretical study ofthemotion
of
an alkylchainattachedtoa
rigid
rod undergoingan anisotropic overallmotion.5The presentworkdealsmainly
with
the dynamicalbe-haviorandtheconformationalpropertiesofpoly(L-lysine)
in
therandomcoilstate by and13CNMR
andrelaxa-tion,i.e.,below
pH
(orpD) 10,wherewell-resolved spectramaybe obtained. Special
attention
hasbeenpaidto therelationship between the nuclear relaxation data, the
proton vicinal
coupling constants, and therotational
isomerismabouteach
of
theC-Cbondsoftheaminobutyl side chains.Asacomplement to the
NMR
studies,ESRexperimentson the spin-labeled polymer provide a straightforward
determination
of
the segmentalmotionof
themain chain in both random coil and-helix
structures. Adirect
comparison
with
proton relaxationdatahasbeenprovided0024-9297/81/2214-0969$01.25/0
bya diamagnetic analogue
of
the spin label.Experimental
Section
Materials. Poly(L-lysine)hasbeen preparedbypolymerization
of L-lysine, the e-amino group being protected by
trifluoro-acetylation. Thisprocedurewas preferred to theoriginalone of Fasmanetal.6becausethe group mustberemovableundermild conditions, particularly in the case ofa spin-labeled polymer. N‘-(Trifluoroacetyl)-L-lysine was prepared from L-lysine and S-ethyltrifluorothioacetateaccording to theprocedureofCalvin et al.7 Conversion to N‘-(trifluoroacetyl)-L-lysine N-carboxy-anhydride(N'-TFA-L-Lys-NCA)was performedby treatmentwith
4Mphosgenesolutionintetrahydrofuran.8 Priorto use NCA was recrystallized from ethyl acetate/petroleum ether. N'-TFA-L-Lys-NCA [2.68g (10"2mol)] was dissolved in25 mL of anhydrousdimethylformamide. Afteradditionof10mg(10"4mol) of n-hexylamine(monomer/initiator ratio=
100),polymerization was allowed to proceed atroom temperatureundercontinuous stirring for2days. Precipitationin100mL ofwater yielded2.0
g(89%) of poly[N'-(trifluoroacetyl)-L-lysine]. Thetrifluoroacetyl group was removed by dissolving0.34 gof
poly[N'-(trifluoro-acetyl)-L-lysine] into 7.5 mL of a 1 M piperidine solution in
methanol. After2h,5mL of1Maqueouspiperidinewas added dropwise understirringtothelattersolution. After2days, the resultingclearsolutionwas dialyzed for5daysagainstcirculating distilledwater at 5 °C andthen againsta 10"3M HC1 aqueous solutionfor2days. Finallythesolutionwas freeze-dried, yielding
205 mg(80%)ofpoly(L-lysine)hydrochlorideas awhite fibrous material.
The spin-labeled poly(L-lysine) (Tempo-PLL)was synthesized following thesame procedure as reported above, replacing
n-hexylamine by 17 mg (10"4 mol) of 4-amino-2,2,6,6-tetra-methylpiperidinyl-N-oxy(Tempo)asinitiator. The diamagnetic
970 Perly, Chevalier, andChachaty Macromolecules H(NH-CH-CO)„-NH (ch2)4 NH3CI" CH3 --S^-CH; Tempo-PLL
analogue of Tempo-PLL was obtained by adding to a 0.1 M solution of PLL 2 equivoffreshly prepared sodiumascorbate
solution. Thereductionofthe Tempo group to
ch3/CH3
N—OH
ch3 ch3
occurredalmost immediately,as checkedby ESR. Theexcess reagentwas removed by dialysis for5daysagainstdistilled water at 5 °C.
The molecular weightsofthe polymerswere determined by measurementoftheintrinsicviscosity of1MNaCl solutionsof
PLLatpH3,6yielding125<DP<145,accordingtothe samples.
The ESR measurementsofradicalconcentration in the
spin-la-beledpoly[TVf-(trifluoroacetyl)-L-lysine] yieldedaDP of146. After removaloftheTFAgroupsweobtainedaDP of138,showingthat
thereisvirtuallyneither chain degradationnor deletionof radical end groupsinthecourse of piperidine treatments. Likewise, the integratedintensity ofthemethylprotons in the diamagnetic reducedformsof Tempo-PLLgivesaDPof140. Sinceallthese
polymers were prepared under identical conditions
([mono-mer]/[initiator] =
100),we mayassume thattheaverage degree of polymerizationof the PLLunder studyis 140,witha com-parativelylow polydispersity.9
NMRand ESRExperiments. ForNMRexperiments,astock
solutionofPLLwas prepared in D20 aftertreatmentat pD7.5
withChelex 100chelating resin toremove metallic impurities. The solutionwas then twice freeze-dried from99.8% D20 at pD
7(pHmeter reading). Beforeuse,PLL hydrochloridewas dis-solved in99.95% or 99.8% D D20for and13Cexperiments,
respectively. AfterpDadjustment the solutionwas flushedwith drynitrogengasintheNMRtube toremove dissolvedoxygen.
AllNMRmeasurementshave beenperformed in the Fourier
transformmodebymeans of Bruker WH90[H13C) = 22.6MHz],
Varían XL-100
M'H)
= 100 MHz], and CAMECATSN-250[y(13C) = 62.86 MHz,i/(*H) = 250MHz] spectrometers.
Thelongitudinalrelaxation times, Tx,were obtained by the inversion-recovery method(180°-t-90°sequences),theinterval
betweeneach sequencebeing atleastequal to5TX. The nuclear
Overhauser enhancements (NOE)were obtained by the inverse gated decoupling method,with long-durationaccumulations to ensure a signal-to-noise rationot less than50 for the spectra
recordedwithoutenhancement. The actualaccuracyoftheNOE
measurementsis±5%. Inallexperiments,theprobetemperature was regulatedwithin ±1 °C.
The ESR experimentson Tempo-PLLwere performedwith aVarían E-109X-bandspectrometer, the magneticfieldbeing calibrated bymeans ofa Varían F-8 magnetometer.
Results
andDiscussion
Motion of the Macromolecular Backbone
andthe
Random Coil
->·«-Helix
Transition.
Theconforma-tional
changesof
poly(amino acids) in solution, inpar-ticular
PLL
andPLGA,have beeninvestigatedbyava-riety
oftechniques,1including NMR.2 The random coil —*· «-helixtransition
isclearlyevidenced byasharpvar-iation
of chemical shifts.3 Similar measurementsperformedon the polymer understudy (DP = 140) gave
us a
transition midpoint
at pD 10.8. Thistransition
has been alsostudied by 13C Tx andchemicalshift
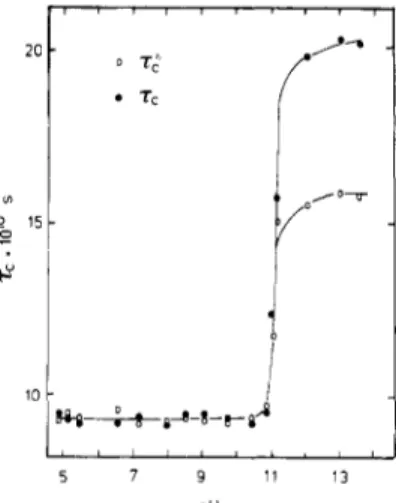
determi-nationsasfunctionsofpH,4butthe broadeningofthelinesFigure1. pHdependenceofthereorientation correlation time of thenitroxideterminalgroup in0.1M Tempo-PLLmeasured
by ESR at5°C.
precluded experimentson the
-helix
formabovepD11.For the measurement of the changein thesegmental
mobility of
themain chainof
PLL
atthe random coil-*-helix
transition,it
seemedto usthat
it
was preferableto performESR experimentson Tempo-PLLupto pD13
on
dilute
(<0.1M)
H20 solutions. AbovepH
11, theprecipitation of PLL
inthe /3-sheetform was avoidedbycooling at 5 °C.10 Undermost
of
our experimentalcon-ditions, the
nitroxide
radical at the chain end gives rise to three equally spaced ESR lines (absorptionfirst
de-rivative),the peak-to-peak
width
of whichdepends upon therelevant nitrogen quantum numbermN= 0,±1.FromAvma, thereorientation correlationtime rcmaybe
obtained bythe relation11
= T2~l(mN)
[3 2/20
+%5( 0)2
+ (b2/8)mN2 -yi5bAyB0mN]Tc +X
(1)with
b = (4tt/3)[A2Zn -y2(AZIN + AyyN)] =2 (
-Aj4)
(2)7
=-{e/h)[gzz
-y2(gxx + gyv)] = (3/3/2ft)(szz-g¡J
(3)In
eq 1, B0,the static magnetic field,is 3250 G,and band
represent theanisotropy of the
A
and g tensors, respectively. AieoN = 47 MHz,AZZN = 99.9 MHz,
giao =
2.0055, andgzz = 2.0022 were readily obtained from the
ESR spectrumof Tempo-PLL inaqueoussolutionatroom
temperatureor frozenat100K.
X
isthecontribution to theelectron transverserelaxationrateTf1,
independent of tc.70,7+,and
L
being the amplitudes of themN = 0, +1,-1 ESRlines (absorption first derivatives),
it
is shown12that
two correlation times may be obtained from the coefficients ofm^ and mN2 in eq 1: 15T2 8b AyB0 AT.sier-te)·]
rifé)"*®"'-]
(4) (5)The differencebetweenrcand rc* isacriterion for the
anisotropy ofthe motion
of
thenitroxide
group.13Figure 1 gives the
pH
dependence of the rc and t*correlation times, which show a steep increase at the
Figure2. (I)13CNOEat22.63MHz
( )
and7\relaxation times at 62.86(·)
and 22.63 MHz (O). The solid lines have beencalculatedforthe Cole-Coledistribution ofcorrelation timestr
(7= 0.7),taking
TR= 7.57X10"16exp(6500 calmol^/RTls. (II) Temperaturedependencesofcorrelation timesfR(dottedline) andtc,thelatterbeing obtainedfromESR linewidths (O)and methyl proton relaxation ofthereducedTempo group
(·).
Thesediagrams are givenfor PLL in the random coil state atpH 7.
H20, instead
of
pD10.8inD20 (meter reading;seeabove), sothat
we assume aspreviously14that
pD a¡ pH.At
roomtemperature below
pH
11,in the random coil form,iso-tropic
motion is observed,with
tc = tc* =* 5 X 10"11 s,whereasa marked anisotropy effectappearsabovepH11,
with
tc 1.5 X 10'9and tc* =* 2 X 10"9s. Thereorien-tationcorrelation timesinthe
-helix
formare significantlyshorterthanexpectedfora
rigid-rod
form, the diffusioncoefficients
of
which are15n 3kTM03
\
i(2V/2Md\
ZflXD±
~2 3 *[2
lis)
MqR /!J
(6) D„ =kTMoJ
6M02R2 ,Z/2V/2Md\l
_4 2
[
Md2
nV3Z
MoR/
\
1M0and
M
are themolecular weightsof
the monomerresidue and
of
the polymer,respectively;M/M0
= 140,d,
the increment
of
the helix lengthper monomer residue, is 1.5Á,R,thehelix radius, is 7.5Á, and , the viscosityof
waterat278K,is 1.512cP. From eq6and7,D± = 6.8X 10sand D\\ = 1.7 X 107 rad s™1. Although and
D±
cannotbe obtained by ESR
in
thepresentcase sincetheorientation
of
theN-0
groupwith
respecttothemacro-molecule backboneisnotknown,
it
isseenthat
tcandt*are much smaller indeedthan2.7X 10"8 s,the valueof (tc)
=
y2(z)|| +
2D±yl.
The ESR measurements on
PLL in
the random coil stateindicateaquasi-isotropic motionof
the chainend,with
tc = t* (Figure 1).This
correlationtime
mayalsobecalculatedfrom the
longitudinal
relaxationof
methyl protons inTempo-PLL
after reduction byascorbic acid. Sincethecorrelationtime fortheaxialreorientation ofCH3 is mostlikely of
the order of 10™11 s or less,it
may beshown16,17
that
the relaxationrateof
aproton pairof
thisgroup is 1
*7 4^
80r6 (3 cos2 -l)2[
2tl 1 +2 2
8rRI
1 + 4o>h2tr2 J (8) where tr isthe correlation time forthe segmentalmotion atthe chain endsand,
the angle betweenaH-H
vectorandpH7,
and therotationaxisofCH3, is90°. Figure2-IIshows
that
thereisan excellentagreement betweenrcandrRwhich
are closetothemean correlation time fRdetermined by
13C relaxation
for
the segmental motionsof
the whole chains. Thelongitudinal
relaxationofa carbonsisgivenby18
l/
—% 7 27 2 2 ~6[1( (
_)
+ 3Ji(cvc) +6«/2( +
)]
(9)The nuclear Overhauser enhancement ofcarbons ob-served under protonnoisedecoupling is18
NOE = ^ + 6»72(^ + wc) ~ Jo(fa>H ~ wc) 7c
=^ (
~)
3e/i(tóc) "b 6«^2( +^c)
In
eq9and10the J(w)are the spectral densitiesexpressedas a
function
of
proton andcarbonLarmor
angularfre-quencies. The13Calongitudinal relaxationandNOEhave
beenmeasured at
different
temperatures atpH
7,wherePLL
isentirely intherandom coilstate.19 Thecomparison of the data obtained at 22.6and 62.86MHz
(Figure 2-1)suggeststhe existenceofadistribution ofcorrelation times represented by a
function
G(tr),
the spectral densitiesbeing expressed as
J(*>)
TrG(tr)
Jo
1 +2 2
E01)
We assume, as in previous works14,20,21 a Cole-Cole
distribution.22 Therefore, the spectral densityisgivenby23
_1__cos
[(1-7)(tt/2)]_
2a> cosh (7 In
)
sin [(1-7)( /2)]
^where 7 is a parameter characterizing the
distribution
width
and tr is the central valueof this
distribution.
The NOEand relaxation time as a
function of
1/T
at the two spectrometer frequencies are consistent
with
7 = 0.7 and tr = 7.56 X 10™15
exp(6500/RT). Thesame
activationenergyof6.5kcalmol-1isfoundinESR,
,
and 13Crelaxationexperiments (Figure2),thecorrelation timeat the chainend given by thetwo former methods being
shorter by only
~12%
thanfR. The motional freedomatchainends is therefore
slightly
higher than the average segmentalmobility
ofthe macromolecular backbone. Here again,therelevant correlationtimefRismuch smallerthan972 Perly, Chevalier, and Chachaty Macromolecules
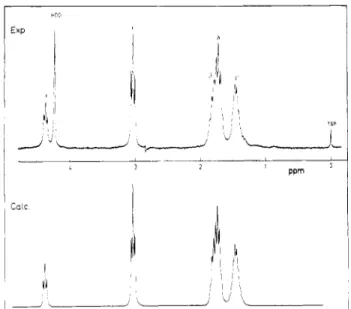
Figure4. Proton NMRspectrum at250MHz, pH= 7,and T = 350K. The lowertracehasbeensimulatedwiththeparameters
ofTable I.
Figure5. Classicalrotamersabout theC-C bondsoftheside
chains.
where is the HCCH dihedralangle.
The populations
of
the classicalT
(trans), G, and G' (gauche)rotamersabout theC-Cbondsof
thesidechains (Figure 5) are related byPT+ PG + PG'
= 1 G5)
TableI
ProtonChemicalShiftsand CouplingConstantsofPLLat 350KandpD 7 Usedin the Simulation of the NMR
Spectrum of PLL (Figure 4) ChemicalShifts”
Hq Hgi Hg, Hy, Hl2 H6||,
HCi~
4.30 1.756 1.827 1.420 1.394 1.673 2.989 CouplingConstants6 Jaa =6.06 JaR = 7.76
Ja"
= 6.50Jal
= 7.704‘
= 7.074e
= 7.56 =Z#3,|32 = ‘/7172= ‘/5152 = ^1e2= -14 “ Inppm from sodium 3-(trimethylsilyl )propionate-2,2,3,3-dt. 6InHz.
expectedfromthe overalldimensions
of
the macromole-cule, the gyration radiusof
whichis(Rg2) =
)
(13)For DP = 140, l= 3.77Á (length ofthemonomer unit),
and
K
= 1.324 one finds(fiG2)1/2 = 50.8 Á. For a
rigid
spherical structureat20°Cinaqueoussolution,one should haveindeedrR= 1.3X 10"7sinsteadoftheobservedvalue
of
5 X 10"10 s.Conformations and Segmental Motions of
Side ChainsatpD
7. Therotamersofthesidechain andtheirinterconversion rateshave beendetermined by and13C
NMR
relaxation. The 250-MHzNMR
spectrumof
PLL
at350K
andpD7isshowninFigure4. Underourexperimental conditions, the Hs and
resonances are
partially
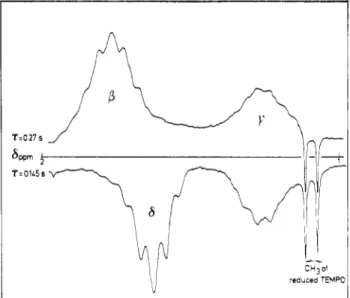
superimposed,givingacomplexpattern,thetwocomponentsof whichhave been separatedby
inversion-recovery (Figure3). Fortime intervals of0.145and0.27
sbetween the180° and90° pulses,theßand resonances are successively canceled, allowing the other one to be selectively observed. The
NMR
spectrumofPLL
has been simulated bymeansof
theSIMEQprogram fromVa-rían, using the parameters given in Table
I
(Figure 4).The proton vicinal couplingconstants have been inter-preted in terms
of
rotamer populations about the C-C bonds,takingsJt= 12.4Hzand3=/g= 3.25Hzasgivenby
Kopple’s modification
of
the Karplusrelation25Vhh
= 1L0cos2 - 1.4cos + 1.6 sin2 (14)
Thecouplings betweenthe a andß protonsyield
PT J«Ai ~ Jg
«W,
(16a) andtherefore Pg =Jt
+J.
-(Jalj ß1+ JaB)Jt-J«
For theß and y protons, we have likewise
Pt
= 'Am 'AmJfJg
ß
JfJ,
Pa' = Jt.-J.
Pr
= ;A2T1 ~ °Jt
-J,
(16b) (17a) (17b) (17c) Thesepopulations maybealsoexpressedasfunctionsofthemean coupling constantsJBiy=
VzCJflm+ and
ß
=VziJ^n
+ J&tr) obtainedfromthesimulationoftheNMR
spectrumof
PLL
(Table I) Pg 2(Jait “ Jg)Jt'Jg
(18a) P<y = 1 -2(Jfl87 ~ Jg)Jt-Jg
(18b) PT = 2(Jai7 + Ja27 -2Jg)4(J8y-Jt)
-7-7.--=
TV
" whereJBy = l/2(Jfsiy +J^)·
The Jgm andJs^2,whichare not directlyavailablefrom
the analysis
of
theNMR
spectrum ofPLL,
may beob-tained by combining eq 17 and 18
^ß ~ — 2^27 (19a) ^0272 ” ~ 2«/^i7 (19b) ^172— ^271 " ~
^av
(19c) where Jav = (Jt+ 2Jg)/3.Table II
Rotamer Populations of PLL Side ChainsObtained from Vicinal CouplingConstants
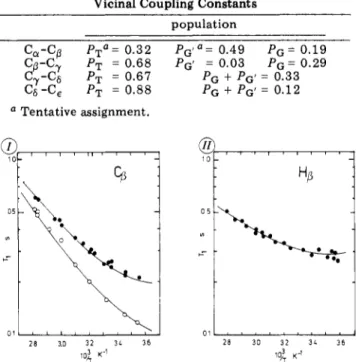
population Ca-C(3
Pt°
= 0.32 Pc'a= 0.49 PG= 0.19 PT = 0.68 PG' = 0.03 Pq= 0.29 C7-Cs Cs-Ce PT = PT = 0.67 0.88 PG + pG'= 0.33 PG + Pg' = 0.12 ° Tentative assignment.Figure6. (I)/3-carbon27\relaxation timesat22.63 (O)and62.86
MHz
(·).
(II) /3-proton T\ relaxationtimes at250 MHz. The solid lineshave beencomputedwithparameters of TableIII. In this figureandthefollowingones the13Crelaxation timesare given astwicethe actual valuesincethe methylenecarbonsare relaxed by the two adjacent protons.For the(CH2)7(CH2)j(CH2)t residue, whereonlyJySand
Jjeare available, the population of the trans rotamer is given byan expression similarto (18c).
It
isseen fromeq15-19that
sincethe measuredvicinal couplingconstantscannotbegenerallyassigned togivenproton pairs,thedetermination of the population
of
the three rotamers is achieved only for the (CH2)s(CH2)r fragment. Inthecase ofthe(CH^CH^
residue,however, the G' rotamerislikelythemostpopulated,26asconfirmedby experimentsinprogresson the paramagnetic relaxation
induced by Gd3+ in
-amino
acids and oligopeptides.Among the rotamers about the
Cr-C{
and Cj-C,bonds,only the population
of
thetrans ones can bedeterminedunambiguously. The populations
of
differentrotamersof thePLL
side chains are given in TableII.
The protonand13Crelaxationtimesinsidechainshave beeninterpretedby assuming
that
rotationaljumps aboutC-Cbondsoccur between threesites,twoofthem, denoted
as 2 and 3, being equiprobable. The motion about the
thesebondsis specifiedby the
jump
rates (site 1 -*site 2 or 3), W2 (site 2 or 3 -* site
1), W3 (site 2 ^ site 3)5,20,27
jn
these calculations,it
is assumedthat
site 1corresponds to the trans conformer
of
theC-C-C-C
fragments.
The populations
of
the sites are given byPi
=V(2u
+ 1)P2 =
P3 =
/(2
+1) (20)
with
=W1/W2; W3hasonlyan influenceon the effective
correlation times governing the relaxation of side-chain nuclei. The expressions
of
the spectraldensitiesinter-vening in eq 9 and 10, whichdepend also on the
distri-bution of
correlation times rR, maybe found inref
20. As pointed out previously,21 the determinationof
WhW2,and W3 hasto bedone by relaxationmeasurements
at
different
spectrometer frequencies and preferablyondifferent
nuclei. We havethereforedeterminedforeachof
the methylene groups the 13C relaxation at 22.6 andFigure7. (I)
2 ,
of13Crat22.63 (O) and62.86MHz(·).
(II) ProtonTx relaxation times at100 (O) and250MHz(·).
The solidlinesare computedwiththeparametersofTable . InpanelIthedottedline corresponds to WJW2= 0.232(parameter
ob-tainedfromVhh).Vf0 = 7 X 1011 s ,andAH= 3.7kcal/mol for
a spectrometer frequencyof62.86 MHz.
Figure8. (I)
2
of13C{at22.63 (O)and62.86MHz(·).
The solid linesare computedwiththe parameters corresponding to modelsIandII ofTableIII. (II) -proton 7\relaxationtimes at 250MHz(·).
Solid line, modelI; dottedline, modelII.Figure 9. (I)
2
relaxationtime ofC,at 22.63 (O)and62.86MHz
(·).
(II)ofH, at100 (O)and250MHz
(·).
Thesolid linesare calculatedwiththe parametersofTableIII formodels IandII.62.86
MHz
as wellas the relaxation at250MHz
and insome cases at100MHz
also(Figure 6-9). The meth-ylene protonsbeing separated by only 1.78Á
(forC-H:
C-H
= 1.09 Á,H-C-H
= 109.5°),their mutual dipolar
interactionisthe mainrelaxationmechanism. The
long-itudinal
relaxationof
theseprotonsisnearly exponential; i.e.,thereisno significant deviation ofthesemilogarithmicplot of
(M0-MZ)/2M0 from the tangent at t = 0. The
contribution of
vicinal protons to the relaxationof
meth-ylene protons, whichisappreciably reducedby therotation
01I_i_i___i_i-1_i_i_i—
28 30 32 3i. 36
974 Perly, Chevalier,andChachaty Macromolecules TableIII
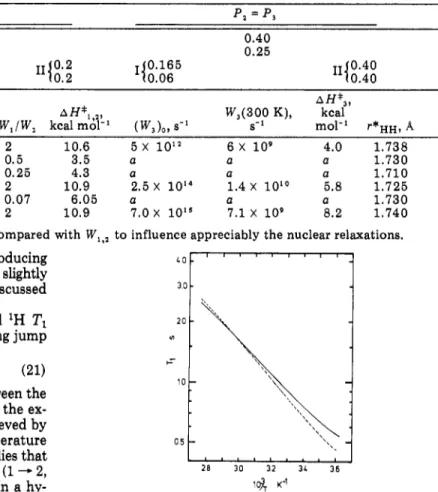
Rotamer PopulationsandKinetic ParametersofPLL Side Chainsfrom ‘H and 13CRelaxationMeasurements P,= P, Ca-Cff Cfl-C-y Cy-Cfi c5-ce f0.67 <0.88 0.2 0.5 II (0.2 10.2 (0.165 <0.06 0.40 0.25 =1;o.4010.40 (W,)o,·-1 W,(300 K),
s"1 WJW, kcal mol"1
# , 2,
(W,)o. s'1W3(300 K), s"1
*
3, kcal mol"1 r*HH, A Ca-Cp 2.5 X 1015 4.4X 107 2 10.6 5 X 10“ 6 X 10’ 4.0 1.738 C0-Cy 5.0 X 10“ 1.4X 109 0.5 3.5 a a a 1.730 Cy-C’S(I) 4.0 X 10“ 2.9X 109 0.25 4.3 a a a 1.710 Cy-CS(II)
1.2 X 10M 1.3 X 10* 2 10.9 2.5X 10'4 1.4X 10“ 5.8 1.725 C8-Ce(I) 1.0 X 10M 3.8X 109 0.07 6.05 a a a 1.730 C8-Ce(II) 1.2 X 10“ 1.3 X 10s 2 10.9 7.0X 10“ 7.1X 109 8.2 1.740The2 3transition rate isprobably too slowcompared with Wli2 to influence appreciably thenuclearrelaxations.
aboutC-Cbonds,was taken
into
accountbyintroducing inour calculationsan effectiveinterprotondistanceslightlysmallerthan 1.78Á. This correctionhasbeendiscussed elsewhere.21
The temperature dependences of the 13C and Tx
relaxation timeshave beensimulated byassuming
jump
ratesof
the formW; =
(W,)0
exp(AHi/RT)
(21)with
i = 1-3,being thepotential barriersbetweenthe
sites (Figures6-10). Agood agreement betweenthe
ex-perimentalandcomputed relaxationratesisachievedby
taking
WJ
W2constant inthe investigated temperaturerange,285 <
T
< 360K. This approximation impliesthat
the differencebetweenthe potential barriersAHX (1-* 2,
3) and AH2 (2, 3 — 1) is
comparatively small.
In
ahy-drocarbon chainAHx
-AH2isindeedoftheorderof0.5-0.6
kcal/mol.24
The populations
Pw
oftherotamersaboutC-Cbondsof
thesidechainas wellas thekineticparametersfor therotational
jumps,deducedfrom the relaxation data,aregiven in Table
III.
Thecalculationshave beenfirst
carriedout by introducing thevaluesof
WJ
W2derivedfromtherotamerpopulations givenby the Kopple’srelationship(eq
14)andadjustingthejumprates W1; W2,and W3
until
themeasured relaxation rates are
fitted with
an accuracybetter than±5% atdifferent temperatures (Figures6-9).
This
process yields a satisfactory agreement for there-laxationsofC9and HdlH6 (Figure6). Onthe otherhand,
the value
of
0.232 givenbyvicinal
couplings for Wx/W2 isnot convenient for therotation
aboutC^-C7. A betteragreement between the observed and computedrelaxation rates
of
Cy,Hw
2
isachievedbytaking Wx/W2 = 0.5.Thisdiscrepancy resultspossiblyfromthe large difference between thepopulations oftheGandG'rotamers(Table
II),
whichcannotbetakenintoaccountinthecalculationsof
relaxationrates. Ontheotherhand, therotamer pop-ulations about C7-CjandCs-C< derivedfrom thevicinal
couplingconstantsseem
fairly
consistentwith
therelax-ationsoftheprotons andcarbonsofthe5-and e-methylene
groups.
It
may be pointed out inparticular that
the conformationof
theC7-C6-C-N
residue is nearly trans andthat
the effectiveH{-H,
distance, whichaccountsfor the proton relaxation, is actuallythat
of the transcon-former
of
analkyl
chain.Several attempts have been made to check whether significantly
different
setsofparameterscan givereason-ablefits
of
thetemperaturedependenceofrelaxationrates. These attempts have not been successful for ß- and-methylene
groups.It
appears, however,that
two setsof parameters are convenient
for
(CH2)j and (CH2)e.Figure10. 2TXrelaxationtimeofC<calculatedforaspectrometer
frequencyof125MHz(H0=11.75T) withthedataof TableIII.
The solid anddottedlines correspond to models I andII,
re-spectively.
One set, designated as
I
in TableIII,
derivedin part
from proton vicinalcouplingsas shownabove,correspondstoan increasingpopulation
of
the localtrans conformer from the macromolecular backbone through the amino groupof
sidechains. The otherset(II)
correspondstoasegmentalmotion occurring by fastexchangeamongthe
two equallypopulatedconformers
with
PT< PG. TheW3jumpratebecomestheneffectiveintherelaxationprocess. Figures8and9showthatthe agreementofmodels
I
andII
withexperimentisequivalent, thecurves computedwith
the correspondingsetsofparametersbeing superimposable except
for
Hs, where a small deviationbetween the ob-served and computed relaxation times is observedfor
model
I
as the temperature increases.It
shouldbeexpectedthat
thediscriminationbetween these two modelscan beperformed bymeansof
aspec-trometer operating atamagnetic
field of
the orderof
10T, which isnow available. Figure10shows
that
thedif-ferencebetweentherelaxation times computedforC{
with
Wx/W2 =* 0.250and2isnot sufficient tomakeanunam-biguous choice between thetwomodels. Model
II
mayberuled out as incompatible
with
the3JHh couplings given in Table I, evenif
eq 14 is notstrictly valid for
the(CH2)7(CH2)6(CH2)efragment.
This
exampleshowsthat
the analysisofvicinalprotoncouplingsintermsofrotamerpopulationsis sometimes essentialin the
interpretation
of
relaxation data.Conclusions
The magnetic resonance study of
PLL
in
aqueoussegmental
mobility
inthe random coilstate and remainsflexibleeven inthe
-helix
form,up to pD 13. Theacti-vationenergyforthe segmentalmotion ofthemain chain isoftheorder
of
6kcal/mol
at pD7,like poly(L-glutamicacid)14andpoly[N5- (3-hydroxypropyl)-L-glutamine]21
un-dersimilar conditions, confirming
that
in therandomcoil statetheflexibility
of polypeptidesisnearly independentof
the nature ofside chains.28The
rotational
isomerism andthejump
rates ofside-chain methylene groupare quite
different
fromthoseof a hydrocarbon chain attached to a macromolecule,29,30 showing, inparticular, for
modelI
ofsegmentalmotion, which seems the most likely, a gradual decreaseof
the reorientationalfreedomfromthemainchainthroughtheterminalgroup.
In
the presentcasethe comparatively slowrotation
of
(CH2)fmaybeexplainedbythe highhydrationdegree
of
the adjacent ND3+group, whichhas been evi-dencedby NMR.31,32Acknowledgment.
Weare greatly indebted to Dr. H.R. Wyssbrod for his
helpful
comments and suggestions concerning theinterpretation of vicinalcoupling constantsin terms
of
rotamer populations. Referencesand
Notes(1) Fasman, G. D. “Poly-Amino Acids”; MarcelDekker: New
York,1967, and referencestherein. (2) Bovey,F.A.Macromol.Rev. 1974,9, 1.
(3Í Bradbury,E. M.; Crime-Robinson,C.;Goldman,H.;Rattle,H. W. E.Biopolymers 1968,6,851.
(4) Saito,H.;Smith,I. C.P.Arch.Biochem.Biophys.1973,158,
154.
(5) Wittebort, R. J.; Szabo, A. J. Chem. Phys. 1978, 69, 1722. (6) Fasman,G.D.; Idelson,M.;Blout,E.R.J.Am. Chem. Soc.
1961,83, 709.
(7) Schallenberg, E.M.; Calvin, M. J. Am. Chem.Soc. 1955, 77, 2779.
(8) Fuller,W. D.; Verlander,M. S.; Goodman, M.Biopolymers
1976, 15, 1869.
(9) Lundberg,R.D.;Doty,P.J.Am. Chem.Soc. 1957, 79,3961. (10) Peggion, E.;Cosani, A.;Terbojevich, M.; Romanin-Jacur,L. J.
Chem. Soc.,Chem. Commun. 1974, 314. (11) Kivelson,D.J.Chem. Phys. 1960, 33, 1094.
(12) Stone,T.J.;Buckman,T.; Nordio,P.L.; McConnell, . M.
Proc.Natl.Acad. Sci. U.S.A.1965,54, 1010.
(13) Goldman,S.A.;Bruno,G.V.;Polnaszek,C.F.;Freed, J. H.J.
Chem. Phys.1972,56, 716.
(14) Tsutsumi, A.;Perly, B.;Forchioni,A.; Chachaty,C.
Macro-molecules1978, 11, 977.
(15) Price,C.;Heatley, F.;Holton,T.J.;Harris,P.A. Chem. Phys.
Lett. 1977, 49, 504.
(16) Navon, G.;Lanir,A. J.Magn.Reson. 1972, 8, 144.
(17) Marshall,A. G.; Schmidt,P. G.;Sykes, B. D.Biochemistry
1972, 11, 3875.
(18) Doddrell,D.;Glushko, V.;Allerhand,A.J.Chem. Phys.1972, 56, 3683.
(19) Myer,Y. P. Macromolecules 1969, 2, 624.
(20) Tsutsumi,A.; Chachaty,C.Macromolecules 1979, 12, 429. (21) Perly, B.; Chachaty,C.;Tsutsumi,A.J.Am. Chem.Soc. 1980,
102, 1521.
(22) Cole,K.S.;Cole, R. H.J.Chem. Phys. 1941,9, 329.
(23) Connor,T.M. Trans.FaradaySoc. 1964, 60, 1574. (24) Flory,P. J.“StatisticalMechanicsofChain Molecules”;
In-terscience: NewYork,1969.
(25) Kopple,K.D.;Wiley,G. R.;Tauke,P.Biopolymers 1973,12,
627.
(26) Fischman, A.J.;Wyssbrod, H.R.;Agosta, W.C.;Cowburn, D. J. Am. Chem.Soc. 1978, 100, 54.
(27) London, R.E.;Avitabile,J.J.Am. Chem.Soc. 1977, 99, 7765. (28) Tonelli,A. E.; Bovey,F.A.Macromolecules 1970, 3, 410. (29) Levy,G.C.;Axelson,D.E.; Schwarz,R.;Hochmann,J.J.Am.
Chem.Soc. 1978, 100, 410.
(30) Ghesquiere, D.;Tsutsumi,A.;Chachaty,C.Macromolecules
1979 12 775
(31) Woodhouse, D.R.;Derbyshire, W.;Lillford,P.J. Magn.Reson. 1975 19 267
(32) Darke, A.;Finer,E. G.Biopolymers 1975, 14, 441.
Conformation of cycle(L-Alanylglycyl-e-aminocaproyl),
aCyclized
Dipeptide Model for
a ßBend.
1.Conformational
Energy
Calculations1®
G.
Némethy,
J. R.McQuie, M.
S.Pottle,
and.
A.Scheraga*lb
Baker Laboratory of Chemistry, Cornell University, Ithaca,New York 14853.
Received August 7,1980
ABSTRACT: The cyclized peptide derivative cyclo(L-alanylglycyl-e-aminocaproyl) contains three peptide groups. Theseare constrained toformaßbendbecausethe distance between the C“ andC‘atomsof the e-aminocaproyl residue cannotexceed 5.04Á,even whenthealkylchainisfullystretched. Therefore,this moleculeserves as amodel compound forbends. Its experimentallyobservedphysicalpropertiescan beused
asstandardsfor the detectionofthe presenceofbendsinpeptides. Ananalysisofthe completeconformational
spaceofthis moleculehasbeencarried out, using energycomputation. The conformationalspaceoftheL-Ala-Gly dipeptidewas mappedinasearchforlow-energy conformationswhich permit ringclosurewiththe e-aminocaproyl
residue. Anumericalsearchmethodwas usedto achieveringclosure. Locallystableconformations were found by energyminimization. Low-energy conformationsoccur only when all three peptide groupsare in the trans conformationbecausethepresenceofeven one cispeptide groupraisesthe energy by at least9.7
kcal/mol. Tenlow-energyconformationsof minimumenergywere found. Twoare typeIIbends,withrelative
energies 0.00and0.93kcal/mol. Fiveare typeIandIII bends,withrelativeenergiesrangingfrom0.74to
1.59kcal/mol. Threeare type and bends,withrelativeenergiesrangingfrom2.80to 3.08kcal/mol.
These resultssuggestthatthe moleculeexistspredominantlyas atypeII bend,withsmall amounts oftype IandIII bendconformationspresent. This predictionwas borneout by experimentalmeasurements in solution and in the solid state (reportedin two accompanying papers).
I. Introduction
Bendsconstituteone
of
theimportant
localconforma-tional
featuresof
proteins, alongwith
a helices andex-tendedchains.2
About
17%of
all dipeptidesequencesinmany proteins ofknown structure occur as bendsor
com-binationsofbends.2,3 The geometricalfeatures
of
bends have been characterized by Venkatachalam,4 who de-scribed andclassified bendconformationsintotypesI,II,
and
III.
Amore general classificationofbendsintoseveraladditional types was introduced byLewis et al.5 They 0024-9297/81/2214-0975$01.25/0 ©1981American Chemical Society