Gas-phase acidity of D-glucose. A density functional theory study
Texte intégral
Figure

Documents relatifs
We perform a KS DFT calculation on the He atom (with the two basis functions given previously) using the standard local-density approximation (LDA) for the
grade II and III meningiomas and when Ki67 labeling index was lower than 10%.Our results suggest that, EGFR protein isoforms without ICD and their corresponding mRNA variants
Table S1 Linear coefficients, c i , in the expansion of the distortion vector, normalized to 1, at the the minimum of 2 A g electronic state in C 2h geometry; correlation of the
This indicated that the nonmagnetic hydrogen and nonmagnetic gold can combine as a magnetic cluster and the Au 7 H n clusters possess tunable magnetic properties by adding even or
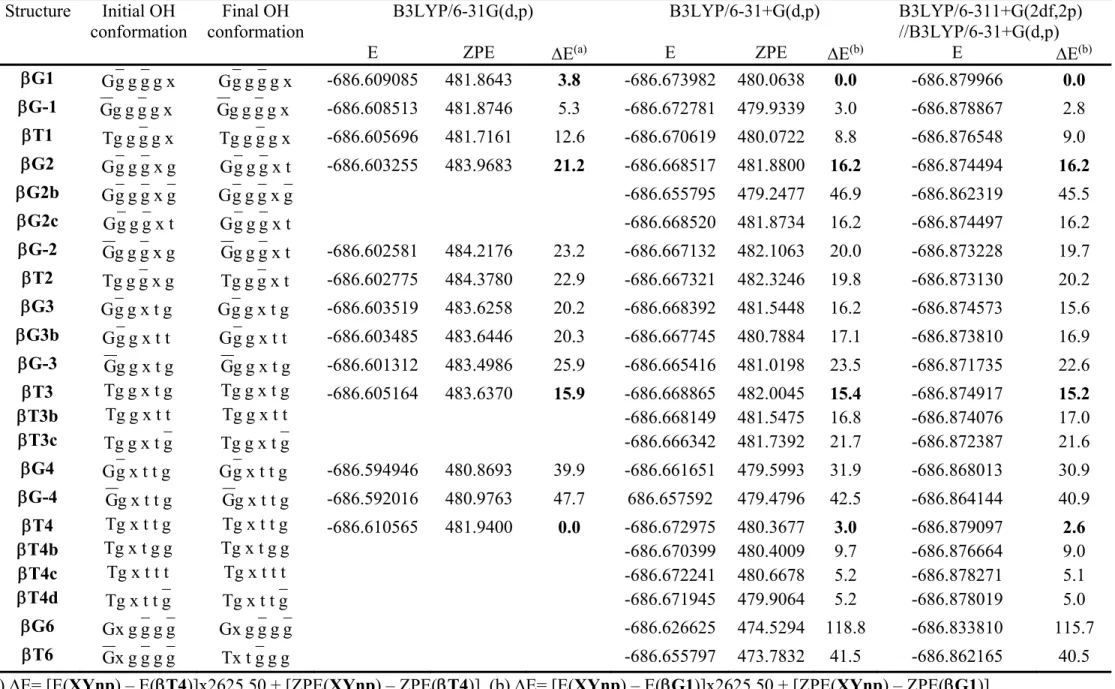
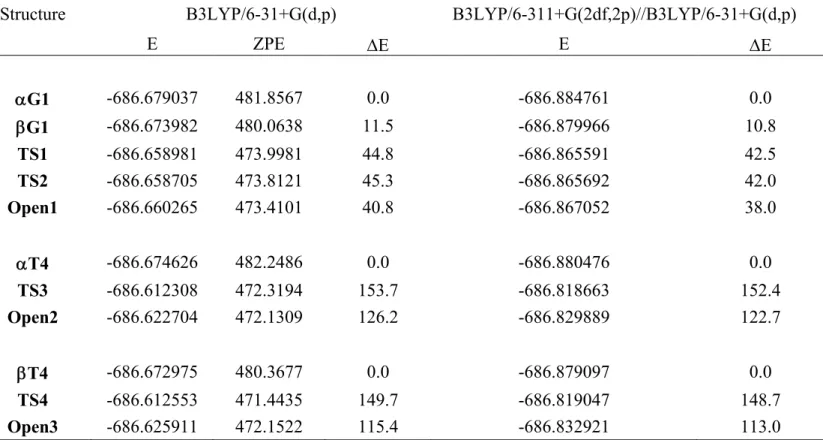
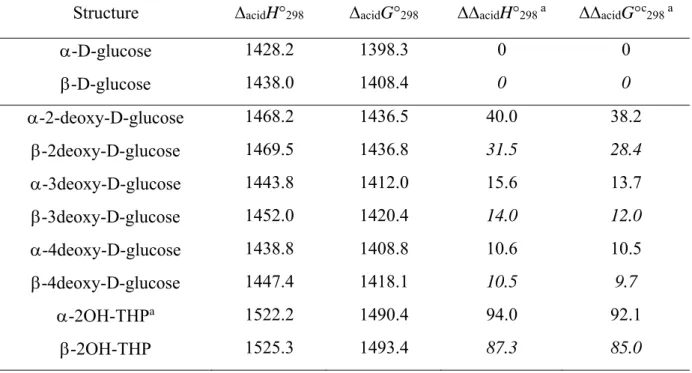
To analyze these results, we have reported in Table 1 the four angles, the nature of the substituents at positions (P) 2, 3, 4, and 5 (P2-P5) assuming that the N-benzyl is at
Considering different initial electronic states of doubly charged uracil, we show that fragmentation depends critically on the energy, shape and intramolecular chemical environment
Solvent equilibrium structure, polarization and beyond Equation 1 states that at the same time as MDFT produces the solvation free energy of arbitrarily complex molecule (the value
When comparing the calculated extremal Fermi-surface orbits with experimental data, we realized a high sensitivity of the computed results on the exact P position within the
