HAL Id: tel-01127522
https://tel.archives-ouvertes.fr/tel-01127522
Submitted on 7 Mar 2015HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Caractérisation de transporteurs membranaires de
Plasmodium falciparum en tant que potentiel cibles
thérapeutiques
Stéphanie Bosne
To cite this version:
Stéphanie Bosne. Caractérisation de transporteurs membranaires de Plasmodium falciparum en tant que potentiel cibles thérapeutiques. Parasitologie. Université Paris Sud - Paris XI, 2014. Français. �NNT : 2014PA11T052�. �tel-01127522�
1
UNIVERSITÉ PARIS-SUD
ÉCOLE DOCTORALE 419 :
BIOSIGNE
Laboratoire : UMR 8221 - Laboratoire des Protéines Membranaires (CEA
Saclay)
THÈSE
SCIENCES DE LA VIE ET DE LA SANTÉ
parStéphanie CORREIA
DE
MATOS DAVID BOSNE
Characterization of Plasmodium falciparum membrane
transporters as potential antimalarial targets
Date de soutenance : 10/10/2014
Composition du jury :
Directeur de thèse : Christine JAXEL Chercheur CNRS, UMR 8221 CNRS, CEA SACLAY
Co-directeur de thèse : Marc le MAIRE Professeur, Université Paris-Sud
Président du jury : Oliver NÜSSE Professeur, Université Paris-Sud
Rapporteurs : Bruno MIROUX Directeur de Recherches INSERM, IBPC, Paris Martin PICARD Chercheur CNRS, CNRS UMR 8015, Paris
Examinateurs : Isabelle FLORENT Professeur MNHN, MNHN Paris Philippe LOISEAU Professeur, Université Paris-Sud
3
Merci à tous ceux qui ont rendu cette thèse possible, rien n’aurait été réalisable sans vous !
A minha Mãe, à Alexandre,
5
Index
Table of Contents
Index ... 5 Table of Contents ... 5 Table of Figures ... 10 Table of Tables... 13 Introduction ... 15 I – Malaria ... 17I.1 – Historical and Sociological Context of Malaria ... 17
I.1.1 – The socio-economic burden of malaria ... 17
I.1.2 – A long story made short ... 17
I.2 - Plasmodium ... 20
I.2.1 – Plasmodium parasites ... 20
I.2.2 – Plasmodium falciparum life cycle ... 21
I.3 – The Disease: Prevention and Treatments ... 23
I.3.1 – The Symptoms ... 23
1.3.2 – Diagnosis ... 23
I.3.3 – Prevention ... 24
I.3.4- The vaccine ... 27
I.3.5 - Antimalarial drugs ... 31
I.3.7 - Antimalarial resistance ... 35
1.3.8 - Finding antimalarials mode of action ... 35
I.4 - New Antimalarial Research... 37
I.4.1 – Strategies for new antimalarial discovery ... 37
I.4.2- New targets explored ... 39
I.4.3 - Biological target validation ... 41
6
I.4.5 – New antimalarials discovered ... 44
I.5 – Artemisinins ... 45
I.5.1 – Artemisinin antimalarial class ... 45
I.5.2 – Artemisinin mode of action ... 47
I.5.3 - Artemisinin resistance ... 51
I.5.4 -Molecular markers of artemisinin resistance ... 52
II – Plasmodium Membrane Transporters as Potential Antimalarial Targets ... 57
II.1 - Malaria Membrane Transport Proteins ... 57
II.2 - Transporters as Potential Drug Targets ... 58
II.3 – Heterologous Expression Systems for Plasmodium Transporters ... 63
II.4 – Generalities on P- ATPases ... 65
II.5 - Generalities on Calcium ATPases ... 67
II.6 – SERCA Pumps ... 69
II.6.1 – Calcium transport ... 69
II.6.2 – SERCA pumps and disease ... 73
II.7 - PfATP6 and Calcium signaling in Plasmodium parasites ... 73
II.7.1– Calcium signaling in Plasmodium ... 73
II.7.2– PfATP6 ... 73
II.7.3– PfATP6 as the proposed target of artemisinins ... 75
II.7.4 – Mutations in PfATP6, responsible for artemisinin resistance ... 78
II.7.5 – PfATP6, is not the direct target of artemisinins ... 80
II.7.6 – PfATP6 polymorphisms ... 83
II.8 - PfATP4 ... 84
II.8.1 – PfATP4 as a proposed Ca2+-ATPase... 84
II.8.2 – PfATP4 as the target of Spiroindolones ... 85
II.8.3 – PfATP4 is a Na+/H+ - ATPase ... 86
II.9 – PfAdT ... 88
II.9.1 – The ATP/ADP carrier family ... 88
II.9.2 – The ATP/ADP carrier in Plasmodium ... 89
II.9.3 – PfAdT as a potential antimalarial target ... 90
III – The project ... 92
7
Chapter I ... 97
Large Scale production of PfATP6 in view of screening of potential antimalarial compounds ... 97
I.1 – Preamble ... 97
I.2 - Article - Antimalarial screening via large-scale purification of Plasmodium falciparum Ca2+ -ATPase 6 and in vitro studies ... 98
I.3 - Cytotoxicity of the Molecules Tested on P. falciparum In vitro Culture ... 110
I.4 – Establishment of an Activity Measurement Protocol in 96-well Microplate and Inhibitor Testing ... 113
I.4.1 – Activity measurement in a 96 well microplate ... 113
I.4.2. – Inhibitors testing on purified PfATP6 ... 116
I.5 – Improvement of PfATP6 Expression and Purification Protocol ... 119
I.5.1 – Improvement of PfATP6 expression protocol ... 122
I.5.2 –PfATP6 purification protocol – thrombin cleavage ... 127
I.5.3 –Improvement of PfATP6 purification protocol – TEV cleavage ... 131
Chapter II ... 135
Expression of PfAdT in view of new inhibitors research ... 135
II. 1 – Preamble ... 135
II.2 – Expression of PfAdT in Yeast ... 136
II.2.1 - Construction of pYeDp60 – His6 – BAD – TEV– PfAdT ... 136
II.2.2 – Expression of PfAdT_wt and PfAdT_K24I in yeast ... 137
II.3 – Expression of PfAdT in E. coli ... 140
II.3.1 –- Construction of pET20b – MBP – Thb – PfAdT ... 140
II.3.2 - Expression of PfAdT_wt and PfAdT_K24I in E. coli ... 141
Chapter III ... 142
Expression of SERCA1a, SERCA-1a_E255L and PfATP6 in Xenopus laevis oocytes and effect of thapsigargin, CPA and artemisinin on the Ca2+-dependent ATPase activity ... 142
III.1 – Preamble ... 142
III.2 - Article - Reappraisal of oocytes experiments on Plasmodium falciparum transporter PfATP6 and SERCA-1a E255L ... 143
III.3 – Conclusions ... 154
8
IV – Perspectives ... 157
IV.I – Perspectives on PfATP6 Studies ... 157
IV.2 – Perspectives on PfAdT Studies... 158
V - Final Conclusions ... 160
Material and Methods ... 163
I – DNA Vector Construction ... 165
I.1 – Yeast Expression Vector Construction ... 165
I.2 – E. coli Expression Vector Construction ... 166
I.3 – Molecular Biology Techniques ... 166
I.3.1 - Polymerase chain reaction ... 166
I.3.2 - Directed mutagenesis ... 167
I.3.3 - Ligation ... 167
I.3.4 – Competent E. coli preparation ... 168
I.3.5 – Transformation ... 169
I.3.6 - Sequencing ... 169
II – Protein Expression, Purification and Activity Measurement ... 170
II.1 – Yeast Strain ... 170
II.2 - Yeast Culture Media ... 170
II.3 - Yeast Transformation ... 171
II.3.1- Verification of the transformation ... 171
II.4- Expression of PfATP6 and PfAdT in Yeast ... 172
II.4.1 - Culture in Fernbach Flasks... 173
II.4.2 - Culture in a Bioreactor ... 173
II.5 - Membrane Preparation ... 175
II.6 - Estimation of Protein Quantity in P3 Membranes ... 175
II. 7 - Estimation of the Quantity of PfATP6 / PfAdT Expressed in P3 Membranes ... 176
II.8 - Batch Purification using BAD domain by streptavidin-Sepharose Chromatography ... 176
II.8.1- Solubilization ... 176
II.8.2 - Fixation to the streptavidin resin ... 178
9
II.8.4 - Protease cleavage ... 178
II.8.5 - Elution ... 179
II.8.6 - Eluate concentration ... 179
II.8.7 - Preparation of TEV protease ... 179
II.9 – Protein Detection ... 180
II.9.1 - SDS-PAGE gel ... 180
II.9.2 - Coomassie blue staining ... 180
II.9.3 - Western Blot... 181
II.10 –ATPase Activity Measurement ... 181
II.10.1 - Coupled enzyme ATPase activity measurement ... 182
II.10.2 - Pi liberation ATPase activity measurement ... 182
II.12 - Expression of PfAdT in E. coli ... 183
II.12.1 - The C43 (DE3) E. coli strain ... 184
II.12.2 -Transformation of C43 (DE3) E. coli strain ... 184
II.12.3 - Expression of PfAdT in C43 (DE3) E. coli strain ... 184
III – Cytotoxic effect of PfATP6 inhibitors ... 185
III.1 – Cytotoxicity Measurement by MTT assay ... 185
IV – SERCA1a, SERCA1a-E255L and PfATP6 Expression in Xenopus laevis oocytes membranes and activity measurement. ... 185
IV.1 – Oocytes Expression Vector Construction ... 186
IV.2 - Expression of SERCA and PfATP6 in X. laevis Oocytes ... 186
IV.2.1 - Phenol-Chloroform DNA extraction ... 186
IV.2.2 – RNA in vitro synthesis ... 186
IV.2.3 – Xenopus oocytes preparation ... 187
IV.2.4 – Microinjection of the RNA into the Xenopus oocytes ... 187
IV.2.5 – membrane preparation from injected oocytes ... 188
IV.3 – ATPase activity measurement of SERCA and PfATP6 and the effect of Artemisinin ... 189
10
Table of Figures
Figure 1 – Spatial distribuition of Malaria endemecity areas in 2010... 18
Figure 2 - Plasmodium falciparum life cycle. A) Schematic representation of P. falciparum life cycle; B) blood smear of infected patients colored with Giemsa. ... 22
Figure 3- Evolution of malaria risk areas from the mid – 19th century until 2010. ... 25
Figure 4 - Prediction of the transmission of P. falciparum according to the climate temperature. ... 26
Figure 5 - Malaria vaccine approaches and targets. ... 28
Figure 6 - Structures of some antimalarials, organized by major chemical family. ... 33
Figure 7 - Timeline of the discovery of new antimalarials and the emergence of the first resistances. ... 34
Figure 8 - Summary of stage-specific activity of the most common antimalarials. ... 36
Figure 9 - Some targets explored for new antimalarial research. ... 39
Figure 10 - Steps for new antimalarial discovery and development. FDA – Food and Drug Administration ... 44
Figure 11 - Artemisinin and Artemisia annua. ... 46
Figure 12 - Potential mode of action of artemisinin. ... 48
Figure 13 - Genomic region of P. falciparum 3D7 strain in chromosome 13 that was highlighted to be associated with artemisinin resistances. ... 56
Figure 14 - Schematic representation of the infected erythrocyte with the membranar complexes highlighted. ... 57
Figure 16 - Phylogeny of P-type ATPase family. ... 67
Figure 17 - Phylogenetic representation of calcium-ATPase transporters in higher animals. sarco-endoplasmic reticulum (SERCA), the Golgi network (SPCA) and the plasma membrane (PMCA) calcium ATPases ... 68
Figure 18 - Schematic representation of the cycle of a SERCA Ca2+ - ATPase. ... 70
Figure 19 - Native structure of SERCA1a expressed in S. cerevisiae. ... 71
Figure 20 - Schematic representation of the structural requirements of SERCA for Ca2+-dependent activation of phosphorylation by ATP. ... 72
Figure 21 - Comparison of SERCA1a crystallographic structure to a structural model of PfATP6. ... 74
11
Figure 23 - Reported inhibition of the ATPase activity of SR and PfATP6, PfATP4 and PfHT expressed in
X. laevis oocytes. ... 76
Figure 24 - Localization of BODIPY-thapsigargin and fluorescent artemisinin, in P. falciparum infected erythrocytes. ... 77
Figure 25 - Inhibition of in vitro growth of yeast K667::pUGpd (blue bars) and K667::pfatp6 (red bars) by different antimalarials. ... 78
Figure 26 - Effect of artemisinin on PfATP6 and SERCA1a expressed in X. laevis. ... 79
Figure 27 – PfATP6 Ca2+ dependent ATPase activity and inhibition by SERCA classical inhibitors. ... 81
Figure 28 - PfATP6 Ca2+ dependent ATPase activity and inhibition by artemisinins. ... 82
Figure 29– PfATP4 Ca2+ - dependent ATPase activity in X. laevis oocytes... 86
Figure 30– Proposed mechanism of spiroindolones on PfATP4 ... 87
Figure 31 – Alignment of human, bovine and P. falciparum mitochondrial AAC. ... 89
Figure 32- Effect of Pcovery compounds on SERCA1a from rabbit muscle. ... 112
Figure 33 - Schematic representation of coupled enzyme ATPase activity measurement. ... 113
Figure 34 - SR and PfATP6 ATPase activities measurement by Pi releasing. ... 114
Figure 35 - Determination of PfATP6 optimal concentration for ATPase activity measurement test by Pi releasing. ... 115
Figure 36 – Effect of CPA on PfATP6 activity. ... 117
Figure 37 - Hybrid antimalarial (4b) based on chloroquine and clotrimazole structures. ... 118
Figure 38 - Effect of a 4-aminoquinoline / clotrimazole-based hybrid compounds on purified PfATP6 and on rabbit sarcoplasmic SERCA1a. ... 118
Figure 39 - Yeast expression vector (pYeDp60) construction with gene of interest. ... 120
Figure 40 - Expression of PfATP6 in yeast. ... 121
Figure 41 - Schematic representation of PfATP6 purification procedure by purification using the BAD domain by streptavidin-Sepharose chromatography. ... 122
Figure 42 - Schematic representation of the protocol performed for the following of the expression of PfATP6. ... 123
Figure 43 - kinetic of the expression of PfATP6 in yeast. Western Blot revealed with an avidin peroxidase probe (Biotinylated proteins). ... 125
Figure 44 - Kinetic of the expression of PfATP6 in yeast. Western Blot revealed with an anti – PfATP6 specific antibody. ... 126
12
Figure 45 - Purification of PfATP6 followed by Western Blot. ... 128
Figure 46 - Purification following and quantification of PfATP6 by SDS-PAGE and Coomassie blue staining. ... 129
Figure 47 - Plasmid construction for pYeDp60 - PfATP6 – BAD, with different protease cleavage sites (thrombin and TEV). ... 131
Figure 48 - Expression of PfATP6 in yeast. Western blot with revealed with avidine peroxidase probe (biotinylated proteins). ... 132
Figure 49 - Purification of PfATP6, comparison of thrombin and TEV cleavage. Western Blot with PfATP6 specific antibody. ... 133
Figure 50 - PfAdT-pYeDp60 vector construction. ... 136
Figure 51 - Expression of PfAdT_wt in yeast. ... 138
Figure 52 - Expression of the mutant PfAdT_K24I in yeast. ... 139
Figure 53 - PfAdT_Wt and PfAdT_K24I in pET20b vector. ... 140
Figure 54 - expression of PfAdT and hAAC in E.coli... 141
Figure 55 - Yeast expression vector pYeDp60 construction with the gene of interest. ... 165
Figure 56 – Design of the direct mutagenesis strategy to obtain PfAdT wt and k24i form of the gene. ... 168
Figure 57 - Yeast growth following in a bioreactor. ... 174
Figure 58 - Schematic representation of the various steps leading to the solubilization of membranar proteins, relatively to free detergent concentration. ... 177
13
Table of Tables
Table 1 - Some targets exploited for vaccine development. ... 29
Table 2 - Some vaccine candidates under development... 30
Table 3 – Main classes of antimalarials. ... 32
Table 4 - Approaches for antimalarial discovery. ... 37
Table 5 - Medicine for Malaria Venture (MMV) requirements for new antimalarials and combination drugs. ... 43
Table 6 – Some of the new antimalarials under development. Source: (Olliaro and Wells, 2009) http://www.malariajournal.com/content/11/1/316/table/T4 ... 45
Table 7 - Some of the most frequently associated candidate gene with artemisinin resistance. ... 55
Table 8-Comparison between the Ki values for artemisinin and thapsigargin inhibition of PfATP6 and SERCA1a and mutants expressed in X. laevis oocytes... 79
Table 9 - Effect of the tested compounds on purified PfATP6, P. falciparum 3D7 and FcB1 strains in vitro growth and cytotoxicity on mammalian Vero cells. ... 110
Table 10 - Medicine for Malaria Venture (MMV) requirements for first stage validated compound hits. ... 111
Table 11 – Comparision of the purification yield between the newly established protocol and the precedent one.. ... 130
Table 12 - Primers used for cloning purposes ... 167
Table 13 - Antibodies used for western blot technique, with the specific proteins they detect and the dilutions at which they were used. ... 181
15
17
I – Malaria
Malaria is a vector-borne infectious disease caused by a parasite of the Plasmodium genus. It is commonly transmitted by a bite from an infected female Anopheles mosquito, which introduces the parasites in the blood stream of a human host. Malaria is a life-threatening disease and half of the world population is at risk of having malaria (World Health Organization, 2013a).
Malaria is endemic from 99 countries in Central and America, Sub-Saharan Africa and South-East Asia (Heppner, 2013; World Health Organization, 2013b) (Figure 1). According to the World Health Organization (WHO) in 2012 there was an estimated of 627 000 malaria related deaths, from about 207 million infected people (World Health Organization, 2013b).
I.1 – Historical and Sociological Context of Malaria
I.1.1 – The socio-economic burden of malaria
Malaria is commonly associated with poverty but it is also a cause of poverty, being an obstacle for economic development. Although it is endemic to tropical regions, it affects travelers from developed countries, affecting people worldwide, beyond borders. It has been pointed to be the major factor for the lag on economic growth retarder in South America. Poverty also increases also the risk of malaria. It is a vicious cycle. In some countries the burden of the disease represents up to 40% of public health spending (http://www.rollbackmalaria.org/index.html). Poverty, political issues, population migration, poor supply and high counterfeit of antimalarial drugs (up to 40% of artesunate-based malaria medications are counterfeited in Asia (Parry, 2005)), heterogeneous access to quality health installations, cultural and religious beliefs, all of these are major factors that contribute to the burden of the disease and to the difficulty of malaria eradication, especially in Africa (Heppner, 2013).
Malaria most severely affects young children and women in their first pregnancy. The major public health problems are: i) the long term cognitive damage in children attained by cerebral malaria, being one of the leading causes of cognitive and developmental disabilities, which largely contributes to the burden of malaria in poor countries (Idro et al., 2005); ii) dual-infection of HIV and malaria (Abu-Raddad et al., 2006; Heppner, 2013); iii) the increased susceptibility to other diseases; iv) and malaria during pregnancy, with lifelong affects to the unborn child (Umbers et al., 2011). Preventing placental malaria in first pregnancy women would prevent one of the great causes of malaria related death and birth of low weight children in sub-Saharan Africa.
I.1.2 – A long story made short
The term Malaria originates from medieval Italian and means mala aria or maus ares, meaning “bad air”. Probably arising from the association of the disease to swamps and marshlands, which we now know are the breading grounds of the mosquitos transmitting the parasites (Reiter, 2000).
18
Figure 1 – Spatial distribuition of Malaria endemecity areas in 2010.
19
I.1.2.1 - The first reports of the disease
Some data suggest that the P. falciparum malaria may have evolved from a gorilla parasite (Prugnolle et al., 2011). Despite the fact that the parasite responsible for malaria has been on earth for 50.000 – 100.000 years, the first references of malaria were in 2700 BC in ancient China (Cox, 2002), but the first evidence of malaria comes from Egyptian mummies of over 5200 years old. The Egyptians also described malaria signs in ancient treaties like the Ebers papyrus, 1500 BC (Cox, 2002). Hippocrates also described clinical signs of malaria, especially fevers, in 700 BC. Through history there exist references to these “periodic fevers”. Malaria was once present in Europe and North America (Figure 3) (Lindemmann, 1999). It was also known as the “Roman Fever” as it could have been at the origin of the decline of the Roman Empire. Malaria dramatically increased due to world population growth and is linked to the development of agriculture and densification of population when the first cities appeared. Agricultural techniques favored mosquito proliferation by increasing breading sites through irrigation runoff and drainage problems.
I.1.2.2 - The first scientific studies
The first important advances in malaria scientific discoveries were made in 1880. Charles Louis Alphonse Laveran was a French doctor of a military hospital in Algeria that observed for the first time parasites in red-blood-cells of an infected patient, and lack of parasites in healthy people. He also observed that the parasites disappeared with the use of quinine (Laveran, 1880). He proposed that this organism was responsible for causing malaria. Laveran called this parasite Oscillaria malariae, a
posteriori it received the common name of Laveran’s germ. One year later a Cuban doctor, Carlos
Finlay, brought strong evidences that mosquitoes were responsible for transmitting the disease, confirming previous suggestions of Josiah C. Nott and Patrick Manson (Tan and Sung, 2008). In 1885 Ettore Marchiafava and Angelo Celli confirmed Laveran’s findings by observing, with an oil immersion microscope, amoeboid parasites in individuals affected by “swamp fever” which they called
Plasmodium. Golgi in 1886, and then Marchiafava and Celli in 1889 described 3 of the 5 Plasmodium
species infecting humans.
The Scottish doctor Sir Ronald Ross described the complete life cycle of the malaria parasite in the mosquito, when working in a hospital of Calcutta (India). In 1897, he dissected infected mosquitoes, and proved that Anopheles was the transmiting vector of malaria to humans. Subsequently he led campaigns for the control of malaria. In 1900 the findings of Sir Ronald Ross and Carlos Finlay were confirmed by a medical committee chaired by Walter Reed, and Ross’s recommendations were implemented in the construction of the Panamá channel, saving thousands of lives and helped to define public health strategies to fight against the disease (Simmons, 1979). In 1902, Sir Ronald Ross received the Nobel price of Medicine. He went on to lead campaigns for the control of malaria. In 1907 Louis Alphonse Laveran also received the Nobel Price of Medicine for his early work on Malaria.
I.1.2.3 – The War
Through history Malaria took an important toll on civilizations. In 1910, Sir Ronald Ross, in his book Prevention of Malaria included a chapter entitled "The Prevention of Malaria in War."
20
During World War I, Cinchona bark and quinine were widely used. The supplies were not enough to cover the needs, in particular because of destroyed cinchona’s plantations. Since then, a lot of financial investments went into new antimalarial research. During World War II and the Vietnam War, malaria was the most important health hazard for the U.S army in South Pacific, infecting 500.000 men (Bray, 2004). In 1942, the U.S.A established the Malaria Control in War Areas (MCWA), and its successor, the Communicable Disease Center, nowadays the Center for Disease Control and Prevention, also known as CDC, was established in 1946. The first chloroquine was synthesized in 1934 by Bayer laboratories, although it was only a decade later that it was commercialized due to its high toxicity (Trager and Jensen, 2005).
I.2 - Plasmodium
I.2.1 – Plasmodium parasites
Plasmodium is a large genus of eukaryote unicellular parasites, also called protozoa. Five Plasmodium
species can infect humans:
Plasmodium falciparum is the most frequent species infecting humans and is responsible for
the most severe form of malaria, the malignant tertian malaria (recrudescent fevers every 2 days). This form of malaria is characterized by severe anemia and can lead to cerebral malaria, a complication that can be fatal. Plasmodium falciparum can be maintained in culture with human erythrocytes. The 23-megabase nuclear genome organized in 14 chromosomes, encodes for about 5,300 genes, and is the most (A/T)-rich genome sequenced to date (Gardner et al., 2002).
P. vivax (typically found in Asia, Latin America, and in some regions of Africa) and P. ovale
(mostly found in Africa and the islands of the western Pacific Ocean) causes a benign form of malaria, the benign tertian malaria. These species have the particularity of developing dormant liver stages (“hypnozoite”). These can relapse several months or even years after the infection has occurred and the disease can become chronic. These people constitute reservoirs of parasites in endemic regions but can also introduce the disease in malaria-free regions. P. vivax is the most widespread species and is responsible for the most prevalent form of malaria it is rarely fatal, but it remains a great cause of morbidity, and thus it is considered a neglected disease (Grellier et al., 2012).
P. malariae (found worldwide) is less frequent and causes the benign quartan malaria (three
days fever cycle). When untreated it can cause a long-lasting, chronic infection that can last a lifetime and cause serious complications. Plasmodium knowlesi is much less frequent and was recently discovered to infect humans in Malaysia (White, 2008a).
21
The only known host of P. falciparum and P. malariae are humans. The other species are zoonotic1 and thus have other animal hosts, such as primate which constitutes a reservoir for the parasites.
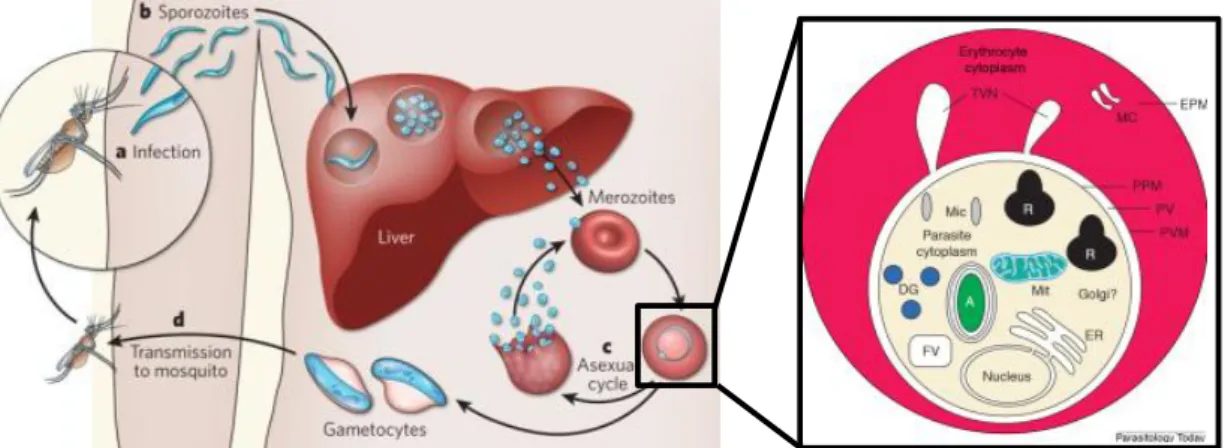
I.2.2 – Plasmodium falciparum life cycle
The Plasmodium falciparum life cycle involves two hosts: a female mosquito vector of Anopheles genus and a Human (Figure 2). Over 30 Anopheles mosquito species are known to transmit human malaria.
A- Infection and Human liver stages - exo-erythrocytic schizogony
P. falciparum lives in the gut and salivary glands of the female Anopheles mosquitoes. During a blood
meal the infected mosquito injects the sporozoites present in the saliva, into the bloodstream of a human (see 1 in Figure 2). Sporozoites migrate and infect hepatocytes (see 2 in Figure 2) where they multiply asexually (5 – 16 days) and mature into schizonts, structure that contains thousands of merozoites (see 3 in Figure 2). During this period no illness is caused (Khan and Lai, 1999). After rupture of the liver cell, the merozoites (see 4 in Figure 2)are free to infect erythrocytes (see 5 in Figure 2) and undergo asexual erythrocytic cycle (see B in Figure 2).
B- Erythrocytic Cycle - erythrocytic schizogony
Each merozoite invades an erythrocyte within 1-2 minutes, consumes the hemoglobin for energy, and becomes an immature trophozoite (ring stage). After maturation and multiplication it differentiates into a schizont (see 6 in Figure 2). The schizonts rupture frees new merozoites that will infect other red blood cells (see 5 in Figure 2) and reiterate the asexual erythrocytic cycle (see B in Figure 2) (every 1 – 3 days). Trophozoits can also differentiate into sexual forms and originate gametocytes (see 7 in Figure 2). The rupture of red blood cells, which liberates merozoites, is responsible for the clinical manifestation of malaria.
C- Sporogonic Cycle - sexual stage
Some of the blood-stage merozoites do not develop into schizonts, but rather differentiate into male and female gametocytes (see 7 in Figure 2). These can be ingested by a female mosquito during a blood meal (see 8 in Figure 2),perpetuating the cycle. The male gametocyte (microgametocytes) and the female gametocyte (macrogametocytes), fertilize in the mosquito’s stomach, producing zygotes (see 9 in Figure 2). The zygotes develop to become elongated - motile ookinetes (see 10 in Figure 2). These penetrate into the mosquito’s mid-gut wall and differentiate into oocysts (see 11 in Figure 2). The oocysts grow, divide and, after 8 – 15 days the oocysts rupture releasing sporozoites (see 12 in Figure 2) that will migrate into the mosquito’s salivary glands.
When this infected Anopheles female mosquito takes another blood meal, it will inoculate the sporozoites present in its salivary glands into another human and the cycle starts all over again. The mosquito acts as a vector, carrying the parasite from a human to another, and is not affected by the
Plasmodium infection.
1
Zoonoses- are infectious diseases that are transmited from a non-human species to a human or vice-versa, with or without an intermediate vector.
2 Parasitemia is a measure of the quantity of parasite present in the blood, reflecting the medical condition of the infection. It can be either expressed in percentage (%) of infected erythrocytes in thin smears of blood, in relation to a enumerated quantity of erythrocytes; or in
22
a
c
b
d
A
B
e
Figure 2 - Plasmodium falciparum life cycle. A) Schematic representation of P. falciparum life cycle; B) blood smear of infected patients colored with Giemsa.
A) sporozoites inoculation; exo-erythrocytic cycle; infected liver cells; schizonts; rupture of schizont, merozoites liberation; erythrocytic cycle; red blood cell infection; schizonts rupture releasing
merozoites; gametocytes (sexual erythrocytic stages); sporogonic cycle (parasite multiplication in the mosquito); mosquito infection during blood meal; male (microgametocytes) and female (macrogametocytes) gametocytes; ookinetes; oocysts, oocyst rupture and sporozoites releasing;
infective stage; diagnostic stages. B) a – schizont; b – mature trophozoite; c – ring stage (immature trophozoite); d – non-infected erythrocyte; e - gametocyte. Source: Centers for Disease Control and Prevention –
CDC, American Society of Hematology, CMAJ. (CDC, NIH,
23
I.3 – The Disease: Prevention and Treatments
I.3.1 – The Symptoms
The liver stages of Plasmodium replication are asymptomatic. Malaria manifests during the erythrocytic cycle of the parasite. The symptoms are: generalized fatigue, loss of appetite, dizziness, intense headaches, digestive problems (upset stomach), nausea, vomiting, joint pain, abdominal pain, diarrhea, diffuse muscle pain. The clinical signs are characterized by: anemia, cyclic violent fevers with intermittent shivers (every 48h, 72h for P. malariae infections, excessive perspiration, convulsions, hemoglobinuria (blood in urine), jaundice, hypoglycemia, splenomegaly and hepatomegaly (spleen and liver enlargement, respectively). Many of these symptoms are non-specific and misdiagnosis becomes a major problem in malaria treatment.
The severe form of malaria appears 6 – 14 days after infection and is almost exclusively due to a P.
falciparum infection (Trampuz et al., 2003). This form of the disease can lead to coma and death.
Pregnant women and under 5 years old children are the most vulnerable. It starts with severe headaches and then cerebral ischemia takes place. In children, the cerebral malaria is characterized by an abnormal posture with the spinal cord in a backwards arching position (opisthotonus position). Severe malaria can progress very fast, in a few days or even hours (Trampuz et al., 2003). A rapid and reliable diagnosis is extremely important to avoid these cases.
1.3.2 – Diagnosis
In many remote malaria endemic regions a simple laboratory test is not possible. Fever periodicity is often used as a diagnosis, although this method is fallible. In endemic regions were antimalarials are widely used either in prophylaxis or for malaria treatment, the evolution of the symptoms is not the same as described above, frequently the rhythmic violent fever periods are absent being rather a gradual fever increase. Malaria is frequently misdiagnosed especially in children, leading to non-adapted treatment.
The most efficient and cheapest diagnosis method is still the microscope observation of a blood smear or a thick blood drop. The advantage of using a thick blood drop is the analysis of a larger amount of blood, increasing the probability of detecting very low parasitemia 2 (down to 0.0001% parasitemia or 5-20 parasites / µl of blood). This technique is often allied to a Giemsa coloring technique (Figure 2 B). This enables a full diagnosis of the disease, by identifying the Plasmodium species causing the infection (Warhurst and Williams, 1996) 3.
2 Parasitemia is a measure of the quantity of parasite present in the blood, reflecting the medical condition of the infection. It can be either expressed in percentage (%) of infected erythrocytes in thin smears of blood, in relation to a enumerated quantity of erythrocytes; or in number of parasites found per µl of a thick drop of blood, in relation to a standard number of red blood cells.
3 Giemsa is a coloring technique first developed by Gustav Giemsa in 1902. It specifically colors chromosomes in a purplish pink with methylene blue and eosin. By visualization of the nucleus of the parasite in the enucleated erythrocyte, parasitemia can be counted.
24
However a microscope and trained medical staff is needed to perform Giemsa technique. In field dispensaries, far from a health structure, it is often not the case. For field diagnosis, rapid diagnosis tests were developed (RDT) (see Box 1) (Abba et al., 2011; Kattenberg et al., 2011). RDT are conceived to be simple to use, with little training required of the health care staff and no special infrastructure needed. These tests are delivered as cassettes, reactive bands or dipsticks and they need only a drop of blood (Pattanasin et al., 2003). They are based on the immune-detection of an antigen from the parasite coupled to a chromatographic reaction. They usually take 15 – 20 minutes to detect the presence of the parasite in the blood sample. The limit of detection of this test is of 100 parasites per µl of blood, whilst a microscope can detect up to 5 parasites by µl of blood.
If a laboratory is available, molecular methods are also possible. By real-time PCR very low parasitemia can be detected (down to 1 parasite per µl of blood) (Mens et al., 2010). This technique is one of the most expensive and requires special conditions, a laboratory equipped with a PCR machine, well trained personnel, and respect of the cold chain during transport and storage of the blood sample. The major disadvantage of these two techniques is that they only show the level of parasitemia of the person, and not the progression of the disease (presence of early or mature stages or even gametocytes); for this the microscopic techniques are very useful.
I.3.3 – Prevention
Malaria prevention measures are the most valuable way to control the spread of the disease. These measures caused a drastic drop of new malaria cases since the beginning of the 20th century, when malaria was present in southern Europe and the United States (Figure 3). Vector control programs largely contributed to this eradication: prohibition of the inundation of agriculture grounds; draining stagnant water, amelioration of sanitation and housing (use of glass windows).
With the new prevention measures implemented by WHO, malaria mortality has been drastically decreasing in the world by 42% since 2000. The WHO prevention goals are: i) the coverage of 100 % of the population with long lasting insecticide-treated nets (ITN), ii) indoor residual spraying (IRS) (see Box 2), iii) protection of individuals by insect repellents and insect repellent clothing, iv) malaria prophylaxis for travelers, v) and vaccine research, along with the regular control of the population and providing treatment to infected people. However, each method has to be adapted to the region
Box 1 | Rapid Diagnosis Tests
The first RTDs developed detected the parasite glutamate deshydrogenase (PGluDH) (Ling et al., 1986), but were replaced by the detection of the lactate deshydrogenase (PLDH). This last enzyme is essential for the generation of ATP during the glycolytic cycle, and hence is one of the most abundant. The presence of this enzyme is narrowly related to a P. falciparum infection. Nowadays there exist numerous RDTs with different specificities and sensibilities, detecting different species of Plasmodium (Ashley et al., 2009; Moody, 2002).
25
under control, for instance, in sub-Saharan Africa some mosquitoes start feeding early in the evening, and in south Asia the vectors prefer being outdoors rather than indoors.
Figure 3- Evolution of malaria risk areas from the mid – 19th century until 2010.
Source: (Mendis et al., 2009; World Health Organization, 2011).
Several initiatives were developed in order to stop the transmission of the disease. The Millenium Developmental Goals form the United Nations (http://www.undp.org/) and is the Malaria Atlas Project, which analysis and maps the spatial distribution of malaria and the climate changes to predict malaria distribution (Figure 4) (Guerra et al., 2007)
Box 2 | Indoor Residual Spraying
Dichlorodiphenyltrichloroethane (DDT) is a molecule known for its insecticidal properties. First synthesized in 1874, its insecticidal action was discovered by the Swiss chemist Paul Hermann Müller in 1939, for which he was later rewarded with the Nobel Prize of Medicine. DDT was then used in the second half of World War II to control malaria and typhus among civilians and troops. DDT turned out to be such a potent insecticide that after the war it was made available for agriculture use, to control plagues at large scale, which led to the emergence of resistant mosquitoes. In the 1960’s the public awareness for the very negative issues of the use of DDT led to the prohibition of its indiscriminate use. Before DDT, in tropical areas of Brazil and Egypt, malaria was controlled by poisoning the natural reproductive habitats of mosquitoes by applying, for instance, Paris Green in stagnant water, which was very toxic. Nowadays other classes of molecules exist such as carbamates and pyrethroids, but DDT remains on of the cheapest insecticide.
26
Figure 4 - Prediction of the transmission of P. falciparum according to the climate temperature.
27
Worldwide researchers have focused their energies in several directions to cover the largest number of possible strategies to discover novel ways to cure/eradicate malaria. These strategies are vaccination development and the discovery of new drugs and new drug targets that haven’t yet suffered from drug pressure. In the fight against malaria, all eyes are turned towards the commercialization of a vaccine that would interrupt the transmission of the disease(World Health Organization, 2013b).
I.3.4- The vaccine
Vaccines are the most cost-effective way to control and prevent a disease. So far a vaccine has never been produced against a parasite. It is particularly difficult to produce a vaccine against malaria due to: i) the variable surface antigens; ii) the genetic variability and the complex life cycle (multistage and multiantigene); iii) the high replication rate; and the rapid evolution of an immune evasive strategy by the parasite (Takala and Plowe, 2009; Takala et al., 2009; Weedall and Conway, 2010). To evade the immune system the parasite has developed astonishing strategies: i) all of the life cycle stages in humans are haploid for rapid selection and transmission of an advantageous mutation; ii) all replication is done intracellularly, to avoid contact with the immune system in this delicate phase and to disseminate rapidly (Mackinnon and Marsh, 2010); iii) merozoites infect red blood cells in less than 30 seconds, leaving little time for the immune system to react (Gilson and Crabb, 2009); iv) and the human immune system imposes a selective pressure that favors the emergence of extensive polymorphism and new antigens (Epstein et al., 2007; Mackinnon and Marsh, 2010). Highly conserved regions have probably not been under this selective pressure, and therefore will be good candidates to the development of a vaccine (Riley and Stewart, 2013).
In the first half of the 20th century, the first attempt to trigger immunization was done by the injection of inactivated sporozoites, the Pasteur vaccine approach (Good, 2013). Unfortunately this attempt was not successful. 30 years later, this approach was revisited: the sporozoites were directly dissected from the mosquito salivary glands, irradiated and cryo-preserved before a subcutaneous injection. This efficient challenge technique in triggering a malaria immune protection by imitating, in a controlled way, the natural route of a human infection (mosquito sporozoite subcutaneous injection) (Epstein et al., 2011; Luke and Hoffman, 2003; Roestenberg et al., 2013; Seder et al., 2013; Vanderberg, 2009). Though, this technique was successful in immunizing people, it was not realistic for a vaccine elaboration. Improvements can be explored because a whole-parasite vaccine has theoretically its advantages, as “all” the parasite proteins are present, reducing the effect of polymorphism in a protein (Good, 2013). Between 1917 and the 1940’s, P. vivax was used as an immune-therapy. The deliberate injection of parasites in healthy volunteers was done in order to obtain an immunization and elaborate a vaccine. This technique was invented by Julius Wagner von Jauregg, for which he received the Nobel Price of Medicine in 1927. However, this technique was very dangerous, killing 15% of healthy people (Vogel and Roberts, 2011).
In the 40’s a success in preventing malaria in primates was achieved by injecting inactivated sporozoites combined with an immune-stimulating adjuvant. Unfortunately this adjuvant was toxic and the sporozoites were obtained from human blood, making this approach impracticable (Clyde et al., 1973a, 1973b; Freund and Thomson, 1948; Nussenzweig et al., 1967; Rieckmann et al., 1974).
28
With the advances of the molecular biology techniques, it was possible to produce recombinant parasites proteins and culture techniques were considerably improved.
There is actually no vaccine commercially available, although almost 20 candidates are being tested in clinical trials (see example in Table 2). Optimism is justified as an acquired immunity against the disease is observed in endemic countries in healthy carriers. Children that survived an episode of severe malaria acquire protective immunoglobulins (IgG) and rarely develop another episode. Immunity against mild malaria is also observed (Cohen et al., 1961). Researches show that if some immunoglobulin is taken from these people and injected into individuals that have no protective immunity, some immunity is acquired (Cohen et al., 1961; Sabchareon et al., 1991).
I.3.4.1 - Potential targets
Figure 5 - Malaria vaccine approaches and targets.
29
The complex life cycle of Plasmodium has been one of the major difficulties encountered in the development of a vaccine. But it is also this complex life cycle that gives so many possible targets and stages to explore (Figure 5 and Table 1).
1. Targeting the very first steps of infection from sporozoite inoculation after mosquito bite to the hepatocyte invasion, or even the infected hepatocyte itself, would confer a “sterile immunity”, i.e. transmission blocking (Duffy et al., 2012; Heppner, 2013).
2. Developing a vaccine against blood-stages like the: erythrocyte invasion, merozoite replication, adherence of erythrocytes to blood vessels walls, or even against infected erythrocytes; could prevent the development of the disease. This would help protecting people from developing symptoms of the disease. Although a rapid emergence of escaping haplotypes can arise from this vaccine (Riley and Stewart, 2013).
3. Designing a vaccine against gametocytes could prevent new infections and hence stop the spreading of malaria. Though this option would not confer any protection to the individual itself, but it will help controlling malaria at a population level. This could be achieved by targeting directly the gametocyte, blocking fertilization or blocking the development in the mosquito mid-gut. The advantage is that gametocytes are poorly polymorphic.
It is important to understand the essential immune effectors, triggering an immune response in humans, to find new antigens that could be exploited as vaccines (Riley and Stewart, 2013).
Targets Mechanisms of action Reference
Protein kinases Are present all through the parasite life cycle (Zhang et al., 2012)
P. falciparum reticulocyte-binding protein homologue 5 (PfRH5)
An essential protein for erythrocyte invasion, with little genetic diversity
(Crosnier et al., 2011; Douglas et al., 2011)
Duffy Binding Protein (DBP) of P. vivax
Binds to the Duffy Antigen (DARC) of erythrocytes during invasion of the red blood cells.
(Batchelor et al., 2014) Variant surface antigens of P.
falciparum PfEMP with a
conserved region of the highly polymorphic VAR2CSA gene
Responsible for adhesion of the infected red blood cells to the placental sulfate A. One was found to be a key target of the immune response, and antibodies against PfEMP1 can confer immunity in children. It is a good candidate to prevent placental malaria
(Chan et al., 2012; Fried and Duffy, 1996; Fried et al., 2013; Salanti et al., 2003)
Table 1 - Some targets exploited for vaccine development.
Perhaps different types of vaccines could be developed, depending on the target population and the region of the globe. We can imagine a vaccine for the populations mostly exposed to P. falciparum, or P. vivax; or a vaccine for travelers to immunize people that were never in contact with the parasite reducing a related public health problem, of cost and side effects of malaria prophylaxis; or developing a tissue-specific vaccine; or adding several antigens in the same vaccine. However these are utopian considerations as no vaccine is yet available.
30
Some vaccines are under development and actually in clinical trials. In the table below we present some examples of these developed vaccines (Table 2).
Vaccine Composition Clinical Trial Phase Reference
SPf66
One of the earliest vaccines developed. It is a synthetic peptide vaccine containing antigens from blood stages linked together with an
antigen from sporozoite stages
Extensively tested in clinical trials in endemic regions but inefficient
(D’Alessandro et al., 1995; Graves and Gelband, 2006)
PfCSP Targets recombinant Circum-Sporozoite Protein (PfCSP)
Did not show any protective immunization during clinical trials, but is
being improved
(Hoffman et al., 1994; Le et al., n.d.; McCoy et al., 2013)
NYVAC-Pf7
Targets multi-stages combining 7 P.
falciparum antigens, from various stages of
the life cycle (sporozoite, liver, erythrocytic and sexual stages).
Cellular immune response of over 90%, in contrast the antibody response was very
low. This vaccine is worth more tests.
(Stoute and Ballou, 1998; Tine et al., 1996)
[NANP]19-5.1ANP
Schizont export protein 5.1 and 19 repeats of the sporozoite surface protein
Successful in conferring immunity in
children and needs more clinical trials (Reber-Liske et al., 1995)
Pfs25 and Pvs25
Malaria transmission blocking vaccine. Antigens conserved between Plasmodium species, interfering with oocytes development
in the mosquito mid-gut
Phase 1 of clinical trials. Dinglasan et al 2008
www.malariavaccine.org
AMA-1/ AS02
Targets blood stages, confers safe and immune protection in adults, however highly
polymorphic and would need to be formulated in a multiallele vaccine
Phase 2 (Ouattara et al., 2013; Thera et al., 2011)
attenuated sporozoite vaccine
Developed for the first time in the 1940’s. The procedure was ameliorated by establishing a
manufacture process to aseptically irradiate and cryopreserve sporozoites.
Under phase 2 clinical trials (NCT01441167). The intravenous route was found more efficient in triggering an immune response. Practical issues are still
pending for delivering intravenously this vaccine on the field, and overcoming the maintenance of a cold chain with liquid
nitrogen
Sanaria Inc, (Alonso and Tanner, 2013; Epstein and Richie, 2013; Epstein et al.,
2011, 2007)
VMP001
P. vivax vaccine. chimeric CSP protein with
repeated regions of two major alleles VK210 and VK247, with AS01 adjuvant
Phase 2 of clinical trials (Yadava et al., 2007)
RTS,S/AS01
Pre-erythrocytic vaccine constituted of a portion of the circumsporozoite protein (PfCSP), the major coat protein of the invasive
sporozoite, which contains B-cell epitopes, and the C-Terminal T-Cell epitopes (T), fused
to the Hepatitis B surface antigen (S), co-purified with additional “S” particles, coupled
to an adjuvant AS01. The CSP antigen is capable of triggering the production of antibodies preventing the invasion of
hepatocytes and destroying infected hepatocytes.
Phase 3
(Alloueche et al., 2003; Bojang et al., 2001; Kester et al., 2009; Regules et al., 2011; Stoute and Ballou, 1998;
Stoute et al., 1997)
Table 2 - Some vaccine candidates under development.
The most recent hybrid subunit recombinant vaccine RTS,S/AS01 is the most advanced candidate that completed phase III of clinical trials in 2011. This vaccine should be available soon and would be used in addition to traditional preventive, diagnostic and treatment measures. The estimated efficacy for children 5 – 17 months age is of 55% reduction of the malaria episodes during the first 12 months, and 47% efficacy against severe malaria. 18 months after immunization the efficacy drops to 46% and 35.5%, respectively. For children aged between 6 – 14 weeks the vaccine is less efficient, reducing cases by only 25% (Agnandji et al., 2012, 2011; Regules et al., 2011; Riley and Stewart, 2013; World Health Organization, 2013c). Although the efficacy and the duration of protection are limited,
31
it is the best candidate nowadays. The RTS,S/AS01 (commercial name: Mosquirix) will be available for African countries with no profit as it is part of the malaria eradication project. It will be available in 2015, regarding approval from the African governments.
Malaria Vaccine Technology Roadmap (funded by Bill and Melinda Gates Foundation) set two major goals for a vaccine with a minimum 50% of efficacy against severe malaria in 2015. A second generation candidate with 80% of prevention of clinical malaria episodes would be needed by 2025 (Heppner, 2013; Heppner et al., 2005; Riley and Stewart, 2013; Vannice et al., 2012) www.malariavaccine.org).
I.3.5 - Antimalarial drugs
The first efficient treatment against malaria was made from the bark of Cinchona tree. This tree grows in the slopes of the Andes, especially in Peru. Indians used it to make a tincture to treat fever. In 1640 it was discovered to be efficient against malaria and was introduced by Jesuits in Europe. Only in 1820 the active ingredient was extracted and isolated from the Cinchona’s bark and identified to be quinine, by two French chemists: Pierre Joseph Pelletier and Joseph Bienaimé Caventou (Kyle and Shampe, 1974).
Quinine became the principal antimalarial treatment until the 1920’s decade. In the 1940’s, quinine is substituted by chloroquine. This new treatment was largely administrated in Southeast Asia and South-America in the 1950’s, and expanded to the entire world in the 1980’s (Achan et al., 2011). Then in the 1970’s artemisinin molecule was discovered from the plant Artemisia annua, and became the most widely administrated drug for malaria treatment (Tu, 2011) (see below section I.5 – Artemisinins, for more information).
Antimalarials used at the present time can be divided into three major classes: Quinolines and derivatives (Hemozoin inhibitors), Antifolates combination drugs, Artemisinin and derivatives (Sesquiterpene lactones) (Grimberg and Mehlotra, 2011) (Table 3, Figure 6). Few of these drugs possess an identified target.
32
Table 3 – Main classes of antimalarials.
Source: (Grimberg and Mehlotra, 2011).
In d iv id u a l d ru gs H emo zo in in h ib ito rs A min o q u in o lin es A mo d iaq u in e, Ch lo ro q u in e, P ri m aq u in e, P ama q u in e 4 -me th an o lq u in o lin es Me flo q u in e, Q u in in e, Q u in id in e O th ers Lu me fant ri n e ( co mb in ed w it h A rt eme th e r), H alo fant ri n e A n ti fo late s DHF R in h ib ito rs Py ri me th amin e, P ro gu an il, Ch lo rp ro gu an il, B ig u an id e s Su lfo n amid es Su lfado xi n e, S u lfam et h o xyp yraz in e Co -fo rmu lati o n Su lfado xi n e/P yri m eth ami n e ( SP) Se sq u it erp e n e lacto n e s A rt eme th er, A rte su n ate , Di h yd ro art emis in in , A rte mo ti l, A rt emis in in O th ers A to vaqu o n e ( w ith P ro gu an il as M alar o n e®) , T etr ac yc lin e, Do xyc yc lin e, Cli n d amyc in , P yro n ari d in e, P ip eraq u in e C o mb in ati o n th e rap ie s Fi xe d -d o se ( co -fo rmu late d ) A rt eme th er/L u me fa n tr in e - A rt es u n ate /A mo d iaq u in e ( A SA Q ) Di h yd ro art emis in in /P ip eraq u in e - A rt es u n at e/P yro n ari d in e A rte su n ate /Me flo q u in e( A SMQ ) O th ers A rt es u n at e/S P - A rt es u n ate /M efl o q u in e - Q u in in e/T etr ac yc lin e Q u in in e/Do xyc yc lin e - Q u in in e/Cli n d amyc in
33
Figure 6 - Structures of some antimalarials, organized by major chemical family.
Some chemical structures of the main classes of antimalarials and other therapeutic and control molecules, are assembled according to either their chemical classes (endoperoxides, 4- and 8- AQs, amino-alcohols), their function (antifolate, antibiotics), or both (e.g., sulfonamides, a chemical class of antibiotic used in combined
34
Chloroquine (CQ) was the safest, cheapest and it is one of the most efficient antimalarial drugs until resistances appeared. However, it is still widely used, especially in combination with other drugs. Antifolate Sulfadoxine/Pyrimethamine (SP) combination drug is one of the other widely used inexpensive antimalarials (Fidock et al., 2004). Amodiaquine and Mefloquine as well as chloroproguanildapsone (LapDap, another antifolate drug) were used after CQ and SP resistances appeared (Fidock et al., 2004). Mefloquine was used during the Vietnam War to treat American troops against multi-resistant malaria. Nowadays it is also used in prophylaxis. Atovaquone is used in combination with Proguanil for travelers malaria prophylaxis, also known under the commercial name of Malarone®. Primaquine is used for the treatment of malaria relapses in P. vivax infections. Antifolate drugs are used in combination with dihydrofolate reductase inhibitors (DHFR) and dihydropteroate synthase inhibitors (DHPS). Artemisinin was first a natural drug isolated from the Chinese herb Qinghaosu - Artemisia annua (Grimberg and Mehlotra, 2011). Proguanil/cycloguanil and pyrimethamine (Peterson et al., 1990), sulfadoxine (Wang et al., 1997), and atovaquone (Srivastava and Vaidya, 1999), respectively target dihydrofolate reductase (DHFR), dihydrofolate synthetase (DHPS) and cytochrome b (McNamara and Winzeler, 2011).
Figure 7 - Timeline of the discovery of new antimalarials and the emergence of the first resistances.
Source: Annex B of "The Race Against Drug Resistance". Mead Over http://www.cgdev.org/blog/antiretroviral-drugs-will-retain-their-power-longer-if-donors-and-gove.
35 I.3.7 - Antimalarial resistance
Some of these drugs have been used for decades and unfortunately parasites have developed resistance mechanisms against some quinolines and antifolates (Grellier et al., 2012; Grimberg and Mehlotra, 2011) (Figure 7). After the increasing death of children under 5 years in sub-Saharan African countries due to chloroquine and sulphadoxine-pyrimethamine resistances emergence, WHO endorsed a policy for using artemisinin-combination therapies (ACTs) as first line treatments, decreasing a lot mortality, along with the introduction of other prevention measures (Dondorp et al., 2010; Korenromp et al., 2003). However this tendency is starting to be reverted with the rising of artemisinin resistant strains in Thai-Cambodian border since 2004 (Alker et al., 2007; Denis et al., 2006; Dondorp et al., 2010; World Health Organization, 2007).It is now more than ever urgent to discover affordable alternatives to the available treatments.
It has been suggested that parasites genetic backgrounds can influence the response to certain antimalarials and the effect of: pfmdr1 (Hunt et al., 2007; Reed et al., 2000; Sidhu et al., 2005) and
pfcrt (Stephanie G Valderramos et al., 2010) on CQ resistance; pfnhe1 on quinine resistance (Briolant
et al., 2011; Cui et al., 2012; Meng et al., 2010).
Resistances in the malaria parasite are particularly prone to emerge due to its genetic and genomic plasticity. It is then important to stay one step ahead of the parasite, in terms of new antimalarials or predicting target mutations to be able to react when such occurs. Though it is not easy and is a very long process.
1.3.8 - Finding antimalarials mode of action
Finding antimalarials mode of action is a rather difficult task and it can be multarget. These can be achieved by several ways:
In vitro induced drug resistance and/or using field resistant isolated parasites, and by
Genome Wide Screening (GWS) try to find mutations only present in resistant parasites. If this is achieved one can learn more about resistant mechanisms or the direct target of the drug (Ariey et al., 2014; Rottmann et al., 2010); another possibility is to demonstrate resistance on a transgenic parasite where the candidate target has been mutated (e.g. the mutated dihydrofolate reductase gene conferred resistance to pyrimethamine (Wu et al., 1996).
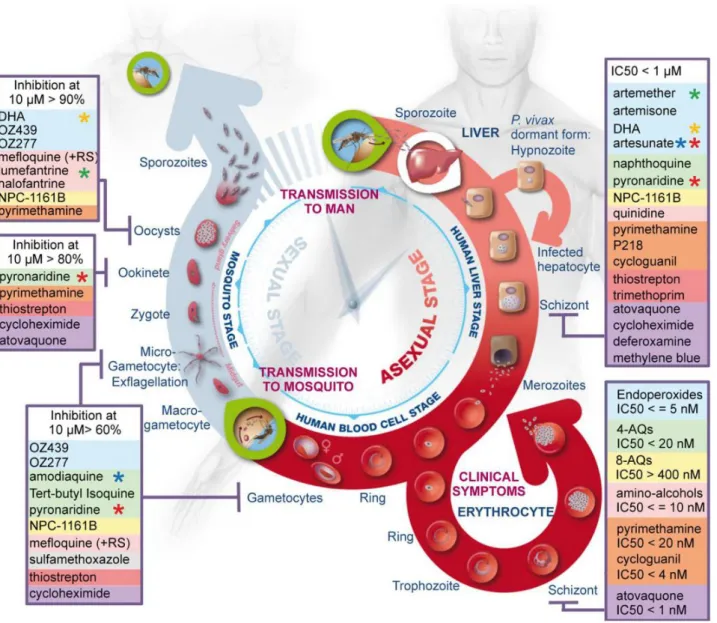
Others focus on trying to find the specific stage of the parasite cycle in which the molecule acts (Delves et al., 2012) (Figure 8), and then try to infer the target by stage specific gene expression profiles.
Another way is by finding specific phenotypes or mechanisms disruption, induced in parasites after drug treatment. One way to test if a molecule with antimalarial activity is targeting a specific pathway or protein, and by repeatedly showing that this molecule has the same effect as a known inhibitor of the target. One can also find a phenotype observable when a
36
protein or a pathway of the parasite is inhibited (swollen food vacuoles of parasites treated with cystein proteases (Rosenthal et al., 2002).
After the target is identified it can be heterologously expressed and purified; a model for a targeted pathway can also be elaborated; P. falciparum genes encoding for drug targets can be expressed in P.
berghei, for which the transfection technology has been developed, and as P. berghei infects mice, an in vivo model is then available.
Figure 8 - Summary of stage-specific activity of the most common antimalarials.
The three major phases, i.e., liver stage, blood stage, and vector stage, of the life cycle of Plasmodium are shown. The two key entry points leading to transmission of the parasites from vector to host and from host to vector are indicated by the green circles. For each developmental stage, the identified actuating drugs are listed
in boxes and coloured as described in Figure 6. Stars indicate components of the main artemisinin combination therapies used: green - coartem; red -pyramax; orange - eurartesim; blue - ASAQ. DHA – dihydroartemisinin.
37
I.4 - New Antimalarial Research
I.4.1 – Strategies for new antimalarial discoveryMalaria has threatened mankind for thousands of years. Even though the prevention initiatives have diminished a lot the number of malaria cases, it is still essential to develop treatments for infected people (especially children and expectant women) and, with the emergence of artemisinin resistance, new antimalarials are urgently needed (Wells and Poll, 2010). Most of the potent antimalarials that we nowadays use were not elaborated with a specific drug target but instead were found from natural products that presented an antimalarial activity (quinine and artemisinin), or semi-synthetic derivatives (chloroquine and artesunate), or even active against other pathogens (antifolates and tetracycline).
Several tools exist that can be exploited for new antimalarials research. Sequencing of P. falciparum genome revolutionized the study of malaria and drug discovery (Gardner et al., 2002) (http://plasmodb.org/) and new tools exist nowadays for the annotation of unknown genes, such as biological data, knowledge charing from the scientific community and the 2007 PlasmoExplore consortium (Florent et al., 2010). Databases have been created such as TDR targets (http://tdrtargets.org) that can be used to predict new targets in silico; or the Plasmodium Protein Data Bank (www.pdb.org) that gathers information about proteins and their structures.
Mainly two global approaches have been defined for antimalarial research:
The screening of molecules issued from chemical libraries either on a phenotypic way, i.e. testing compounds upon in vitro cultures of Plasmodium (whole cell approach) (Table 4); Searching for inhibitory molecules that act upon the biochemical activity of a potentially
identified drug target that can be an essential enzyme or pathway, ideally specific to the parasite (target based approach) (Chatterjee and Yeung, 2012) (Table 4);
Target-based approach Cell-based approach (whole cell)
Characteristics Identification of a target from bibliography, orthology with targets form other organisms, genomic data
Genetic and chemical validation of the target Target expression and eventually purification
Development of a high-throughput compound screening test on the target
Compound testing in vitro on Plasmodium, cytotoxicity tests on mammalian cells, in vivo testing
Compound optimization
Identification of libraries of potential antimalarial molecules
High-throughput compound screening test in vitro on
Plasmodium, cytotoxicity tests on mammalian cells, and in vivo testing
Compound optimization Pros Known mechanism of action, possibility of optimization with
target knowledge
Active on Plasmodium (cell penetration), the target can be later identified by several tools Cons Difficulties in expression and eventual purification of the target;
genetic and chemical target validation required, development of a specific high-throughtput screening test required
Mechanism of action unknown, optimization can be difficult
Table 4 - Approaches for antimalarial discovery.
38
But other strategies to find new antimalarials exist such as: (Grellier et al., 2012) New drug design from the knowledge of a target;
The optimization of already existing antimalarials by drug combination, rearrangements/modification of existing compounds in order to limit the side effects of these;
Multi-target hybrid antimalarial design or new combinations of already existing antimalarials (polypharmacology), overcoming antimalarial resistance issues when only one target is aimed (Morphy and Rankovic, 2005);
Adaptation of drugs used to cure other pathogens or diseases such as cancer; exploring natural products;
Or even target validation upon orthologs of other Apicomplexan parasites that present evolutionary relation and are technically easier to manipulate, like for example Toxoplasma
gondii.
Although it is not required to know the target of an antimalarial, it is rather useful because if this molecule fails clinical trials it is always possible to explore this target in future compound research, and also to predict potential resistances (McNamara and Winzeler, 2011).
Going backwards and identifying the unknown targets of established antimalarials can also be useful. These can be then exploited to design and test new molecules with more affinity to the target, or by targeting a different binding site of the same validated target, and hence exploring a region that has not suffered from drug pressure. For example, CQ, that was discovered to interfere with hemoglobin degradation pathway, this is now a validated target explored for other antimalarials (Biagini et al., 2003; Ursos and Roepe, 2002). Structural knowledge of the target will help to design new antimalarials, exploiting the regions non- homologous to host proteins.
Common sense tells us that a future good target should be a feature absent in humans (e.g.: the prokaryotic apicoplaste), or an ortholog with very different characteristic. In order to avoid toxicity and to be able to specifically target the Plasmodium feature. The only difficulty is validating the target from no previous information. If one can exploit the percentage of difference between these two orthologues and extract molecules acting specifically on Plasmodium, then there is no danger in using orthologues proteins. There are even advantages in this procedure that rely on the eventual knowledge of the human orthologue. A validated target in another organism (human or other pathogen) than Plasmodium can also be exploited (Fidock et al., 2004). Some examples of this strategy is the targeting of cysteine proteases by a cysteine protease cathepsin K, which is a treatment against osteoporosis (Rosenthal et al., 2002; Rotella, 2002), or targeting the farnestyl transferases inhibited by molecules against the human orthologue used in cancer treatment (Figure 9) (Chakrabarti et al., 2002; Gelb and Hol, 2002).
39
Figure 9 - Some targets explored for new antimalarial research.
Adapted from (Fidock et al., 2004).
I.4.2- New targets explored
Some examples of the targets explored are cited below. Drug discovery strategies that are focused upon exploring new targets have a vast choice of targets that can go from physiological/metabolic pathways to the study of single proteins such as transporters and kinases and pathways such as the haeme polymerization mechanism (Grellier et al., 2012; Grimberg and Mehlotra, 2011).
i) Transporters are integral membrane proteins and enable the transport of solutes across biological membranes. They are promising targets as they are generally involved in essential pathways and resistant mechanisms, such as PfCRT, Pgh1 and PfNHE1. Some transporters have been localized and some have been heterologously expressed as recombinant proteins.
ii) Parasites lipidome and lipid synthesis (Maréchal et al., 2011) are good drug targets because, post invasion of the host cell, innumerous membranes are synthethized during rapid cellular