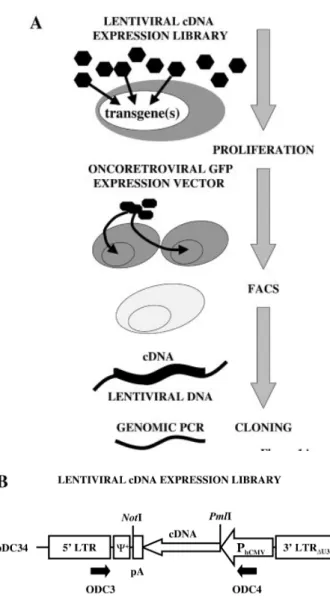
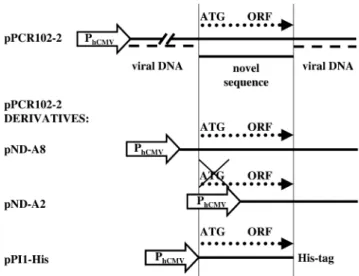
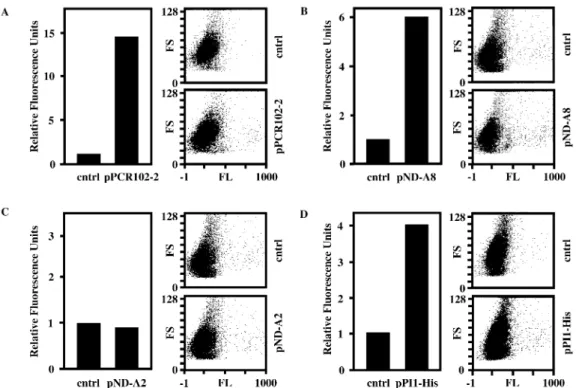
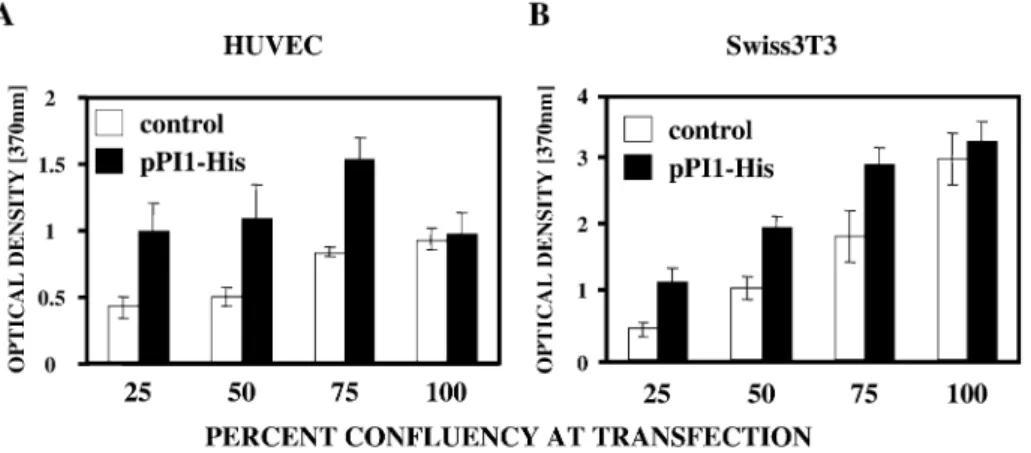
Identification of a novel proliferation‐inducing determinant using lentiviral expression cloning
7
0
0
Texte intégral
Figure
Documents relatifs