Is it possible to detect G×E interactions in GWAS when causal exposure is unobserved?
10
0
0
Texte intégral
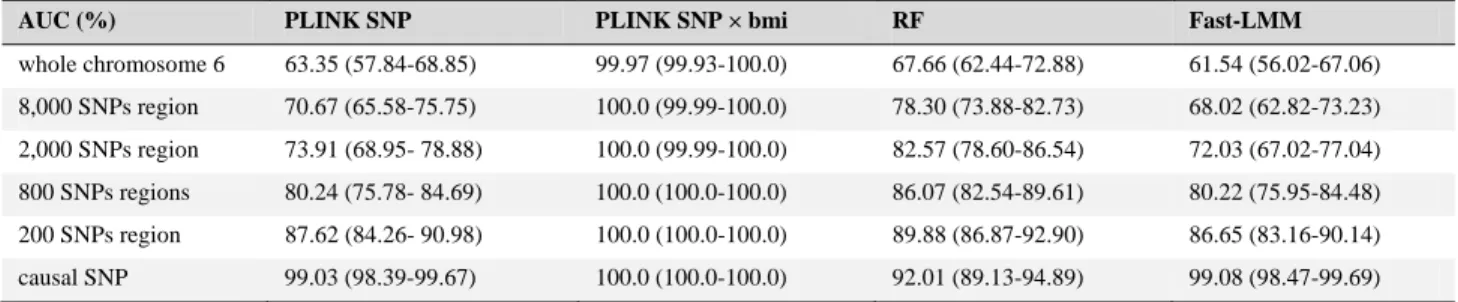
Figure


Documents relatifs