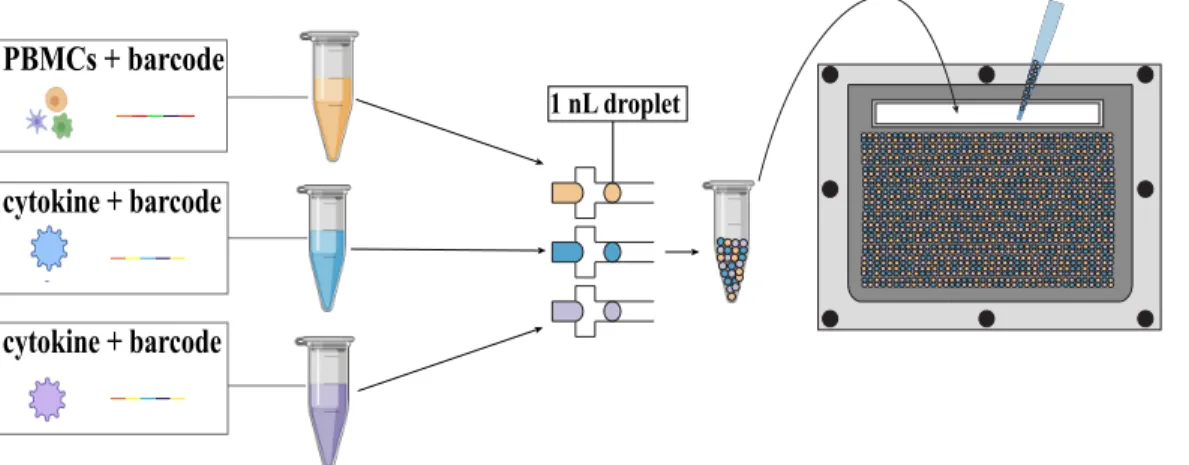
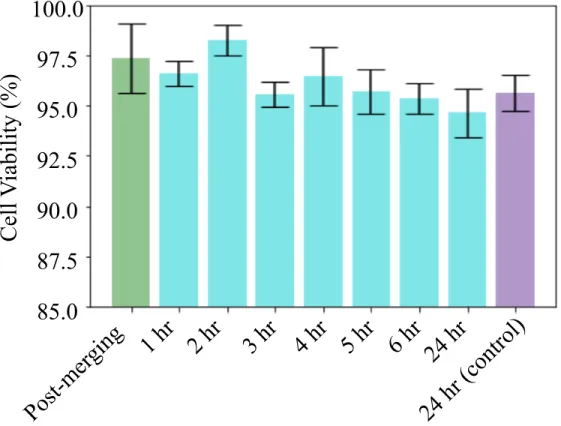
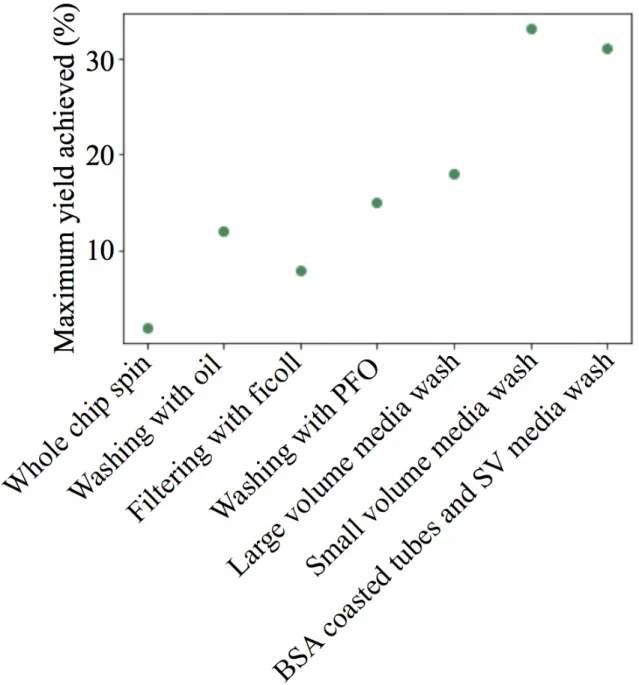
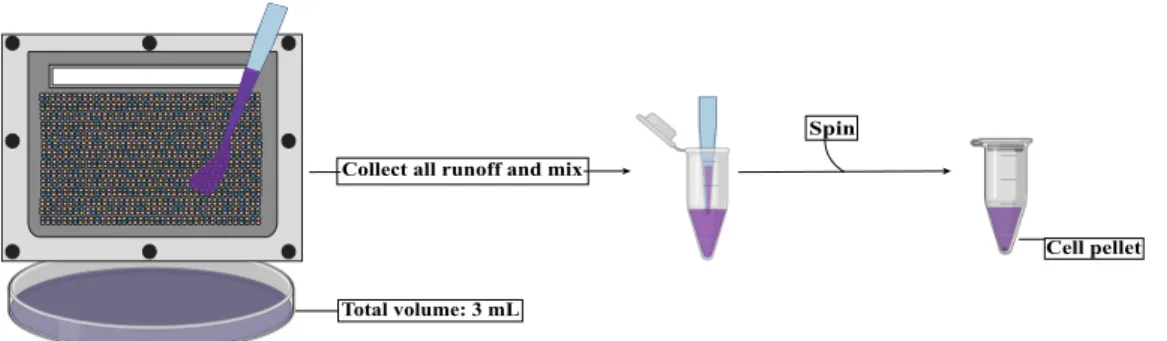
Development of a microfluidic droplet system for immune Cell multiplexing experimentation
55
0
0
Texte intégral
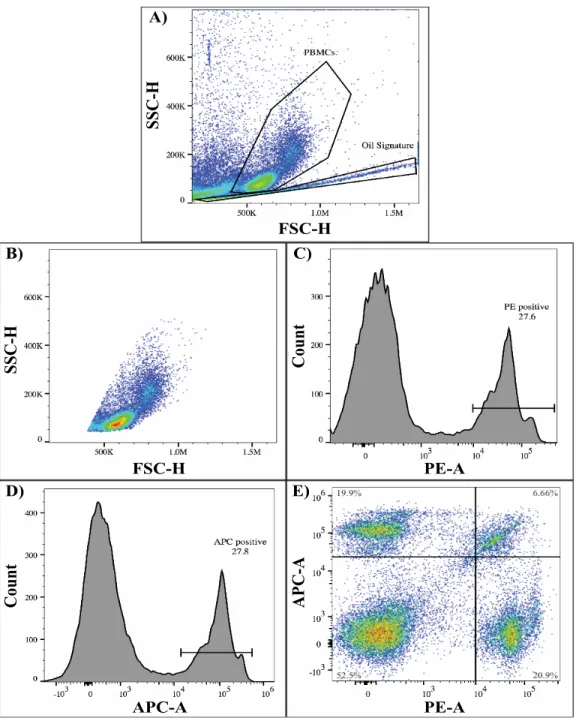
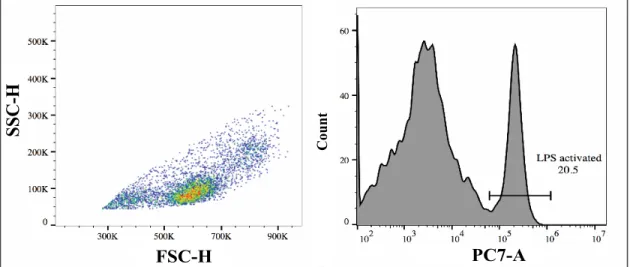
Figure



+7

Documents relatifs