Ab initio calculation of the rotational spectrum of methane vibrational ground state
Texte intégral
Figure
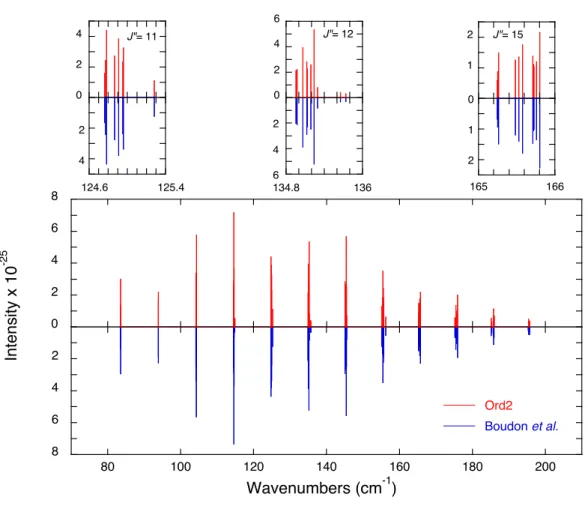
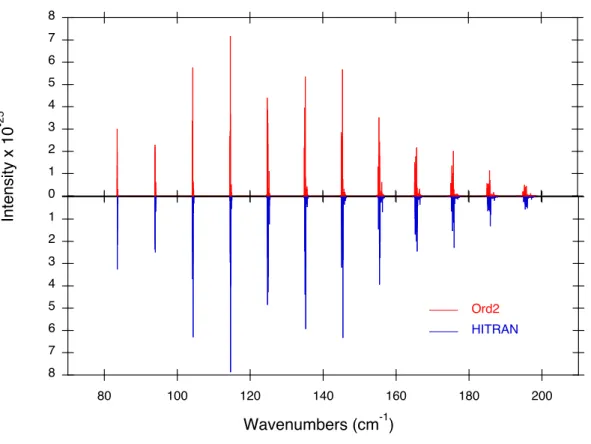
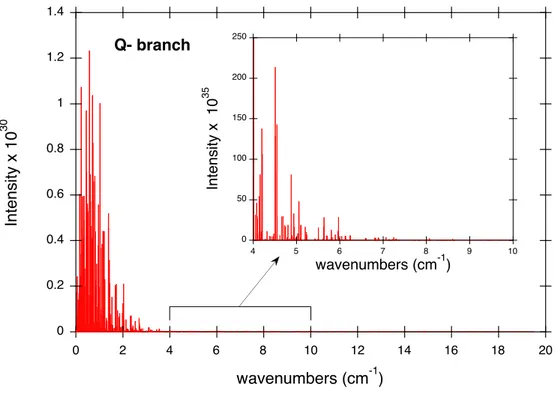
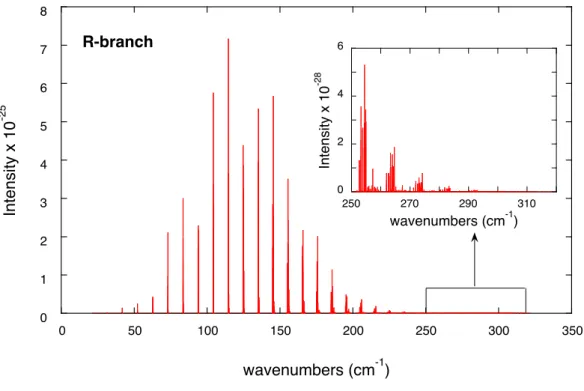
![Fig. 3. Relative errors of the calculated transition wave numbers with respect to those deter- deter-mined by STDS [80] and of calculated intensities with respect to those obtained at SOLEIL](https://thumb-eu.123doks.com/thumbv2/123doknet/13097534.385792/43.892.215.640.151.832/relative-calculated-transition-numbers-respect-calculated-intensities-obtained.webp)
Documents relatifs
We have added into the new MIRS a possibility for the user to generate a list of symmetry allowed terms involved in the ITO normal mode expansion of either an effective
potential we have obtained the phonon dispersion curve using the self-consistent harmonic approximation and compared results with experiment.. To obtain the interionic
The Bertlmann-Martin inequality [1, 2] yields an upper bound to the ground state mean square radius (ms radius) from the lowest dipole excitation energy.. It concerns a particle
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des
3 ىلإ لولأا ثحبملا يف قرطتن لاكشلإا اذى ىمع ةباجلإل " ضورفملا رعسلا رظح " مث ، ىلإ يناثلا بمطملا يف قرطتن " ضورفملا رعسلا رظح ءازج ."
The low-lying vibrational states of H 2 CO are determined and compared to harmonic, quartic force field correlation-corrected vibrational self-consistent field (cc-VSCF), 40
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des
Abstract - An application of the molecular dynamics simulated annealing method for performing total energy calculations is made to the study of the micro- scopic structure of
![[PDF] Cours Production de code Assembleur en PDF | Formation informatique](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)