Publisher’s version / Version de l'éditeur:
Vous avez des questions? Nous pouvons vous aider. Pour communiquer directement avec un auteur, consultez la
première page de la revue dans laquelle son article a été publié afin de trouver ses coordonnées. Si vous n’arrivez pas à les repérer, communiquez avec nous à [email protected].
Questions? Contact the NRC Publications Archive team at
[email protected]. If you wish to email the authors directly, please see the first page of the publication for their contact information.
https://publications-cnrc.canada.ca/fra/droits
L’accès à ce site Web et l’utilisation de son contenu sont assujettis aux conditions présentées dans le site LISEZ CES CONDITIONS ATTENTIVEMENT AVANT D’UTILISER CE SITE WEB.
The Journal of Physical Chemistry C, 124, 18, pp. 10079-10084, 2020-04-13
READ THESE TERMS AND CONDITIONS CAREFULLY BEFORE USING THIS WEBSITE. https://nrc-publications.canada.ca/eng/copyright
NRC Publications Archive Record / Notice des Archives des publications du CNRC : https://nrc-publications.canada.ca/eng/view/object/?id=b5967c34-1063-4b7e-9304-ca8c5dcf88d0 https://publications-cnrc.canada.ca/fra/voir/objet/?id=b5967c34-1063-4b7e-9304-ca8c5dcf88d0
NRC Publications Archive
Archives des publications du CNRC
This publication could be one of several versions: author’s original, accepted manuscript or the publisher’s version. / La version de cette publication peut être l’une des suivantes : la version prépublication de l’auteur, la version acceptée du manuscrit ou la version de l’éditeur.
For the publisher’s version, please access the DOI link below./ Pour consulter la version de l’éditeur, utilisez le lien DOI ci-dessous.
https://doi.org/10.1021/acs.jpcc.0c03101
Access and use of this website and the material on it are subject to the Terms and Conditions set forth at
Development of Fukui function based descriptors for a machine learning study of CO₂ reduction
Development of Fukui Function based descriptors for ML study of CO
2reduction
S. Siahrostami, A.Tchagang, S. Stoyanov, S. GusarovAbstract
We propose a new type of Machine-Learning descriptors based on Fukui function (FF) and its following projection to the Connolly surface formed by smoothed vdW spheres of individual atoms. The FF contains an information (chemical activity) about the local system’s response to perturbation and so it might replace the computationally expensive mapping of adsorption energy of small molecules as an indicator of catalytic activity. We show that FF supplemented by general characteristic of electronic structure of the surface like work function, is well correlated with the topology of mapped adsorption energy. We also discussed different possible ways of implementations are discussing.
Introduction
Heterogenous catalysts are the key component of the chemical and solar energy production industry. The surface of the catalysts significantly promotes the reaction rate by facilitating the bond breaking of reactants and bond formation in products. Adsorption of intermediates on the catalyst surface is a key to lead to useful changes in reactant bonds and following formation of intermediate pre-product complexes. The optimum catalyst must adsorb reactants and releases the products rapidly, to avoid blocking the active sites. This phenomenon is known as Sabatier principle, which states the interactions between the catalyst and the adsorbates should be "just right", that is, neither too strong nor too weak. Unfortunately, no precise instruction on how to determine the “optimal adsorption strength” is given and so the Sabatier principle can be treated as qualitative only. Understanding and especially predicting energy differences associated with bond-formation and bond-breaking reactions occurring on the surface of solid materials and nanoparticles is the central problem in heterogeneous catalysis [1].
Initially, the development of catalysts was based on an empirical approach complemented by the chemical intuition. Later, the number of theories was proposed to go beyond the Sabatier principle and make the discovery of catalysts more sustainable [2]. However, the real revolution was done by density functional theory (DFT), calculations using DFT become a powerful tool for studying catalytic processes and helped to identify new and improved catalysts [3]. In a series of pioneering works, Nørskov [4-6] and co-workers suggested a way to understand the chemisorption of reaction adsorbates in terms of the electronic structure of the catalyst (so-called d-band theory, relating binding energies to the electronic structure of the surface represented by the position of d-band center calculated by DFT approach). This enables mapping of parameters determining the rate of a catalytic reaction onto a reduced phase space spanned by energy parameters known as descriptors. In the d-band model, the band of catalyst d-states participating in the interaction is represented by a single state with energy Ed (d-band limit of the
Newns-Anderson model), also known as the center of the d-band. The change in the adsorption energy correlates the shift of Ed: a stronger upward shift indicates the possibility of the formation
of a larger number of unoccupied anti-bonding states, leading to the higher binding energy. On the other hand, lowering Ed leads to weaker bonding, so the position of d-band center can be considered as an indicator/descriptor of the reaction. Hammer-Nørskov model successfully explains both the experimental and the first-principles theoretical results for different heterogeneous catalytic processes. In the subsequent studies, the position of the catalyst d-band center was also correlated with the adsorption, activation and dissociation energies of small molecules (like H, CO, etc.) which provides a simple and efficient precursor to many chemical processes (Fig. A in attachment). This model was successfully generalized to model catalytic activity of the magnetically polarized transition metal surfaces [7]. It was also confirmed in numerous experimental and theoretical reports that modeling of the adsorption of simple molecules can give an insight that is difficult to obtain from experiments alone but needed for sustainable chemical production, alternative energy solutions, and pollution mitigation.
One of such perspective applications is the development of efficient catalysts for the conversion carbon dioxide (CO2) to chemicals and fuels. Coupled with renewable energy sources (like wind or solar) this reaction is an appealing way to generate carbon-neutral fuels or industrial chemicals and also an attractive method for storing intermittent renewable energy. However, together with the increase of industrial demands, this requires the development of new technologies and solutions, and, overall, next generation of catalysts are needed? based on the latest achievements in the material science allowing fine-tuning of reactivity (nano defects, porous structure, dopants, alloys, etc.). In turn, this requires a way to quickly model the catalytic activity of a large number of potential systems due to variations of possible probing systems (e.g. different facets and terminations of bimetallic alloy compositions). For example, a simple screening of catalytic activity of Nickel-Gallium alloys represented by four bulk compositions (Ni, NiGa, Ni3Ga, Ni5Ga3) and forty surface facets (with Miller indexes less or equal 3) results in 583 possible adsorption sites and requires more than 70000 single point DFT calculations [8]. This leads to the necessity of the development of a new paradigm to model novel catalysts which could be achieved through artificial intelligence (AI) and machine learning (ML) framework. Modern ML methods can fit complex functions in high-dimensional feature spaces while selecting the most relevant descriptors automatically by learning the highly complicated relationships between the independent and dependent variables via non-linear “black box” data processing. Recent advances in AI and ML offer the potential to enhance Material Science research broadly. In particular, ML shows a great promise for the design of new materials and processes for materials synthesis, and also the development of methodologies for the discovery process itself [9]. In contrast to traditional computational techniques utilizing heavy quantum chemical calculations, the machine-learning platform teaches itself by seeing more and more examples using flexible algorithms. At its core, the AI approach offers the possibility of training computers with the provided catalytic properties of materials and evaluates/predicts the top potential catalysts for a reaction of interest [10].
Nevertheless, despite its great success, there are important challenges in the deployment of an intelligent framework for designing new catalytic materials. Presumably, this is due to the complex nature of catalytic processes and the stringent requirements of ideal catalysts, which should be highly efficient, environmentally benign, stable under working conditions and made
of earth-abundant elements. As it was outlined before, the huge number of potential candidates make it almost impossible to apply DFT to map the activity of catalyst surfaces with the energetic parameters (e.g. binding energies) because it requires calculations that can be costly for large scale screening of materials. This has motivated researchers to find an alternative way for describing the activity of the catalysts based on their local geometric and individual atomic features of the active sites. The number of the generalized coordination numbers (e.g. typical valence, number of bonds, ionic radius, etc.) has been proposed instead of DFT energetic descriptors [11]. However, although these descriptors, called atomic fingerprints, could be successfully used to predict physical (thermodynamics, viscosity, boiling point, fracture toughness, density, etc.) properties of bulk materials, the modeling of catalytic reactions requires chemical properties (spectroscopy, excited states, chemical activity, band gaps, orbital energies, work functions, etc.) and related to transition states search which is one of the most difficult and computationally demanding tasks in quantum chemistry [12].
In the proposed study we aim to develop new descriptors which will provide a balance between accuracy and computational cost and also form a basis for the development of the new ML approach to study catalysis. In particular, they should capture important physicochemical properties of surfaces, be much easier to compute than the current DFT-descriptors (e.g. binding energies), and uniquely define each system. Then machine learning tool will be developed and used to find patterns, build models, or discover descriptors that map the features describing the catalyst to their figures of merit.
Method
The concept of the Fukui function was introduced between 1952-1954 years by K. Fukui and coworkers [13,14], as a reaction of the electronic density of the molecular system to adding or removing of some number of electrons. A decade later, it was developed further by Parr and Yang [15,16]. They point out the strong connection of FF with chemical activity and catalytic properties. Functionally, this quantity is defined as the derivative of the electron density, (r) with respect to the number of electrons of the system N (at constant external potential v(r), defined by positions of nuclei and external fields acting on molecular system), alternatively, it could be also expressed as a variational derivative of the chemical potential, , with respect to the external potential 𝑣(𝑟⃗) at constant number of electrons, N [15]:
f(𝑟⃗) = (∂ρ(𝑟⃗)
∂ N )𝑣(𝑟⃗) = ( δμ
δv(r⃗⃗))𝑁. (1)
In other words, it expresses the sensitivity of the chemical potential of a quantum system to an external perturbation represented by 𝛿𝑣(𝑟⃗). In theory of metals, it is also possible to connect FF with the density of states (DOS) [16,17]:
f(r⃗) =g(Ef,r⃗⃗) g(Ef) (2) where 𝑔(𝐸𝑓, 𝑟⃗) is the local density of states (LDOS):
g(ϵ, r⃗) = ∑ |ψ(r⃗)|2δ(ϵ i− ϵ) i = 2V (2π)3∫ 𝑑𝑘⃗⃗|ϕ𝑘(𝑟⃗)| 2δ(ϵ(𝑘⃗⃗) − ϵ), (3)
This justifies the use of the Fukui function concept to describe the catalytic reactivity of metal and metal-oxide surfaces.
While the reaction, in general, is governed by the principle of electronegativity equalization [21] which assumes the balancing of chemical potentials (negative of electronegativity, =-) of reactants through redistribution of electronic density, the local reorganization is determined by electrophilic and nucleophilic Fukui functions (as the number of electrons, N is a discrete variable, the right and the left derivatives of 𝜌(𝑟⃗) with respect to N are discontinuous):
𝑓−(𝑟⃗) ≡ (∂ ρ ∂ 𝑁) , 𝑓
+(𝑟⃗) ≡ (∂ ρ
∂ 𝑁) (4)
The chemical potential and electronegativity are global properties of reactants. Electrons tends to move from reactant with high potential to reactant with low potential in order to balance the resulting value. The FF characterizes a local ability of molecular system to donate/accept electrons. For example, regions with large positive values of f + are most favorable for nucleophilic
attack while regions with negative values of f - is measured by the change upon subtraction of an
electron and denotes the most suitable site for electrophilic attack. An interplay of nucleophilic and electrophilic FF is defined by ratio of chemical potentials of reactants [17, 20]. In case of large difference in chemical potentials of reactants, the reactant with higher potential value will transfer a number of electrons (N>0) from regions where its f - is large to the regions with large
values of f + of the reactant with lower number of chemical potentials. On the other hand, if values
of chemical potentials are similar, the reactants equally donate and accept electrons and true derivative needed to describe density redistribution:
𝑓0(𝑟⃗) ≅𝑓+(𝑟⃗)+𝑓−(𝑟⃗) 2 (5)
This also implies that an appropriate combination of global and local parameters is especially important for construction of the accurate and meaningful ML descriptors in order to map the most important features of heterogeneous catalysis and to avoid overfitting by using semi-supervised like learning algorithms. For example, using only local activity indicators could result in erroneous and overcomplicated correlation between 𝑓± and chemical activity. With the right choice of descriptors, local parameters define a general way for reaction, while the local parameters specify the particular interaction and following redistribution of electronic density which help to compare different sites in different molecular systems (as it was pointed out in [22]).
On the other hand, the modern ML algorithms allow to take nonlinear associations and higher‐ order interactions into account automatically. Therefore, in this study we use the FF because it is a general and well-studied reactivity index, in contrast to recently introduced activity indexes such as Fermi local softness or dual reactivity descriptors [26]. The ML algorithms could resolve the complex relations of FF and reactivity.
To better represent the surface reactivity, we project 3D FF onto the molecular surface formed by rolling a ball over the Van der Waals surface. This approach has a number of advantages compare to projection of FF to atomic sites, so-called condensed Fukui function obtained by approximate integrations of the Fukui function over atomic regions. First of all, the mapping to 2D surface allows to better take into account different relative orientations/positions of reactants (on-top, bridge, hollow, etc.). This allows to investigate all possible active sites, including vacancies and nanostructured defects. Second, the surface distribution allows to accurately represent the topology [23] of the surface and its differential features. Moreover, to better fit a specific reaction, one can convolve the 2D surface projected function with typical patterns representing local area for that reaction. This method could be used to model different stages of reactions and/or complex interactions with large molecular systems involving a number of space-separated covalent bonds. In the following studies we will compare the performance of FF with other local descriptors.
In order to determine the best distance to the reference surface, we run a number of calculations to study the change of FF with the distance for a number of different surfaces (Ni, NiGa alloys). Figure 1. represents one of such illustrative examples. It is clearly seen that meaningful interaction starts from the distances approximately equivalent to VdW atomic radii. This also agrees well with the study of an electrophilic/nucleophilic attacks which are featured by a small electrostatic force at the reaction center on its van der Waals surface descriptors of nucleophilic and electrophilic regioselectivity [24].
Lastly, as it was pointed out by J. Jiang et al. [25], the choice of global descriptors is challenging because many parameters have their own limitations. For example, due to many possible reaction pathways the chemical potential cannot be clearly defined in some cases. Therefore, to distinguish different types of catalytic surfaces we use work function which is also widely used as a global descriptor for photocatalyst/catalyst design.
Results
To illustrate the proposed concept, we have studied the correlation between FF projected onto the Connolly surface and CO binding energies mapped to the surface, as main precursor of catalytic activity. The initial calculations were done with DMol3 package with DNP and PBE basis set [] and the rest of calculation were performed within periodical DFT with norm-conserving pseudopotentials and pseudo-atomic localized basis functions as implemented in the OpenMX software package. The conventional generalized gradient approximation (GGA) was chosen for the exchange-correlation functional with the PBE density functional with D2 dispersion correction.
The double-ζ polarized (DZP) basis set with
We investigated 12 surfaces of Ni, NiGa formed by different slabs with most frequently used combination of Miller indexes. For each surface two type of calculations were performed.
First, to map binding energies of we run a set of distinct geometry optimizations for chemisorption of CO molecule along the lines parallel to z-axis which is perpendicular to the slab (Fig. 2a). The coordinates of that lines form a rectangular grid in xy-plane. Contour plots of binding energies are presented on Fig 2a.
The second type involved the calculation of FF for bare surfaces by finite field method constructed from electronic densities of neutral and electron/hole doped systems (q=0.5). Then, the values of FF were approximated on Connolly surface (Fig. 2b) for the points corresponding to tabulated map of binding energies.
Next, we perform linear regression analysis of FF projected onto surface and calculated 2D mapped binding energies (Fig. 3). It is clearly seen that there is a strong correlation between these two quantities. On Fig 3a we present a regression for Ni (100) surface. Contour plots for these calculations has the same structure and correlates well. However, for a combined set of different slabs, results have a visible deviation which can be significantly decreased by complementing FF quantities with work functions. This shows an importance of supplementing of local distribution by global quantity defining the reaction globally.
Conclusions
The development of efficient and accurate computational ML models to study surface catalysis face with serious challenges related to the nonlocal character of interaction of compounds with the surface as well as with the multistage nature of the process. These two factors make it difficult to utilize the descriptors which are typically used for modelling physical properties and simple chemical reactions. For example, the applicability of electronegativity, unusual and typical valence, etc. are restricted to the local active sites on the surface and require some additional adjustments when used to model the bridge-bonding to two different atomic sites. The multistep character of catalysis, on the other hand, suppose the different character of functional dependence on different steps which complicates the machine learning process.
In the present work, we suggest the new descriptors for ML study of catalysis on the surface based on projected Fukui function and its further convolution with probing radial basis function (ML) to better represent the functional dependence for the particular step of reaction.
In the present work, we suggest a new type of descriptors to better represent the catalytic surface in a machine learning procedure. It is based on projected Fukui functions which reflects the response of the molecular system for an electrophilic/nucleophilic attack. In this approach the local quantity should be supported by global parameter(s) characterizing the chemical activity of surface in general. The principal advantage of introduced descriptors is that the Fukui function projected to an accessible surface provide the local chemical activity of the system that can be compared to the ML generated pattern functions corresponding to each step of the reaction. This model could be successfully applied to study periodical and nanostructured catalytic surfaces and will allow to select the active sites on whole surface.
The concept might be improved further by convolution of 2D surface distribution of FF with specific local patterns for particular reaction (and/or reaction stages). This will be a subject of our next developments. Also, we are planning to implement a two-step algorithm for more accurate predictions of catalytic activity. On the first step we will use the general projected FF database of catalytic surfaces to select potentially active surfaces and their catalytic centers. Next, for the number of selected systems we will enhance the database by direct DFT calculations of activity (e.g. binding energies) to improve the procedure.
Acknowledgements
NRC program AI4D and Clean energy
References
1. A.J. Medford, A. Vojvodic, J.S. Hummelshøj, J. Voss, F. Abild-Pedersen, F. Studt, T. Bligaard, A. Nilsson, J.K. Nørskov, From the Sabatier principle to a predictive theory of transition-metal heterogeneous catalysis J Catal, 328 (2015), pp. 36-42
2. J. Wisniak, The History of Catalysis. From the Beginning to Nobel Prizes, Educ. quím., 21(1), 60-69, 2010
3. Alejandro E. Pérez and Rafael Ribadeneira, Modeling with DFT and Chemical Descriptors Approach for the Development of Catalytic Alloys for PEMFCs,
http://dx.doi.org/10.5772/intechopen.80922
4. Hammer, B. & Nørskov, J. K. Electronic factors determining the reactivity of metal surfaces. Surface Science 343, 211–220 (1995).
5. Hammer, B. & Nørskov, J. K. Why gold is the noblest of all the metals. Nature, 376, 238–240 (1995).
6. Hammer, B. & Nørskov, J. K. Theoretical surface science and catalysis—calculations and concepts. Advanes in catalysis, 45, 71–129 (2000).
7. S. Bhattacharjee, U. Waghmare, S. Lee, An improved d-band model of the catalytic activity of magnetic transition metal surfaces, Scientific Reports, 6:35916, DOI: 10.1038/srep35916 8. ACS Catalysis 2017, 7, 6600
9. Y. Liu,T. Zhao, W.Ju, S. Shi, Materials discovery and design using machine learning, Journal of Materiomics, v3(3), 2017, Pages 159-177
10. B. Goldsmith, J. Esterhuizen, J. Liu, C. Bartel, C. Sutton, Machine Learning for Heterogeneous Catalyst Design and Discovery, AIChE Journal, 2018 Vol. 64, No. 7, p. 2311-2323
11. J. Noh, S. Back, J. Kima, Y. Jung, Active learning with non-ab initio input features toward efficient CO2 reduction catalysts, Chem. Sci., 2018,9, 5152-5159
12. Z. Li, S. Wang and H. Xin, Toward artificial intelligence in catalysis, Nature Catalysis, v1, 2018, pp641–642
13. K. Fukui, T. Yonezawa, H. Shingu, A molecular orbital theory of reactivity in aromatic hydrocarbons, Journal of Chemical Physics, 20 (1952), p. 722
14. Fukui, T. Yonezawa, C. Nagata, and H. Shingu, ibid. 22, 1433 ~1954! 15. Parr RG, Yang WT (1984) J Am Chem Soc 106: 4049
16. W. Yang and R. G. Parr, Hardness, softness, and the Fukui function in the electronic theory of metals and catalysis, PNAS, 1985, 82(20), 6723-6726
17. Allison, T.C., Tong, Y.J. (2012). Application of the condensed Fukui function to predict reactivity in core–shell transition metal nanoparticles. Electrochimica Acta, Volume 101, page 334-340.
18. X. Wang, G. Zhang, L. Yang, E. Sharman, J. Jiang, Material descriptors for photocatalyst/catalyst design, Comput. Mol. Sci., 2018, 8, p1369
19. P. Chattaraj, A. Cedillo, and R. Parr, Fukui function from a gradient expansion formula, and estimate of hardness and covalent radius for an atom, J. Chem. Phys. 103, 10621 (1995)
20. Parr RG, Donnelly RA, Levy M, Palke WE (1978) J Chem Phys 68: 3801
21. Sanderson, R. T. Chemical Bonds and Bond Energy; Academic Press: New York, 1976.
22. Gurkan, Y. Y., Turkten, N., Hatipoglu, A., & Cinar, Z. (2012). Chemical Engineering Journal, 184, 113–124,
23. Falivene, L., Cao, Z., Petta, A. et al. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 11, 872–879 (2019)
24. Phys. Chem. Chem. Phys., 2017, 19, 1496
25.
X. Wang, G. Zhang, L. Yang, E. Sharman, J. Jiang, Wiley Interdiscip. Rev.:
Comput. Mol. Sci. 2018, 8, 1369
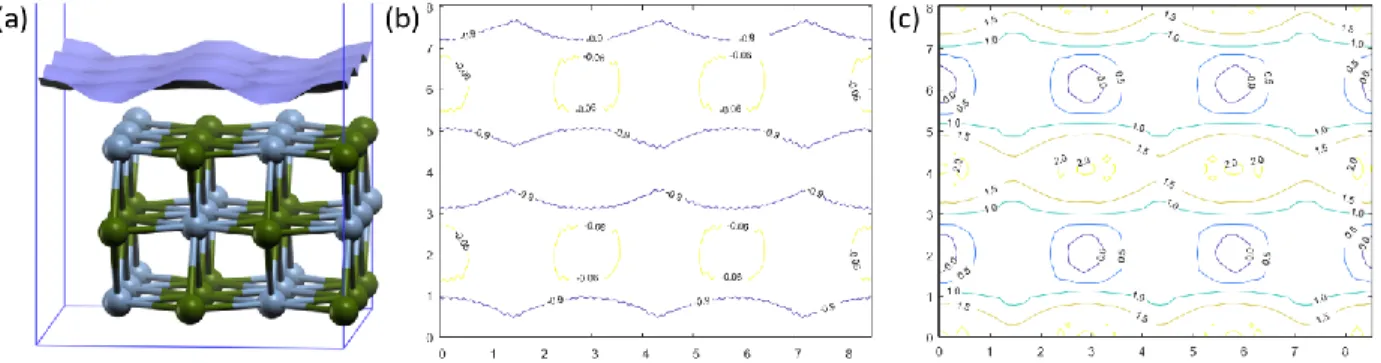
Figure 1: The redistribution of nucleophilic (b) electrophilic (c) Fukui functions for different distances between CO molecule and Ni (110) surface
Figure 2: The top (a) and side (b) views of NiGa (110) surface with positions of chemisorbed CO molecules (each molecule corresponds to a separate calculation of chemisorption of single CO, as for example, CO represented by ball and sticks which can move along z-axis); contour plot (c) of mapped binding energies and its 3D representation (d)
Figure 3: Connolly surface for NiGa (110) (a) and contour plots of projected nucleophilic (b) electrophilic (c) Fukui functions
Figure 4: Linear regression of projected Fukui function and mapped binding energies of CO molecule for Ni((110) surface (a); for set of 12 studied surfaces (b) and its modification for multivariable linear regression in case when FF is augmented by work function (c).