Comprehensive Proteomic Analysis of Human Erythropoiesis
Texte intégral
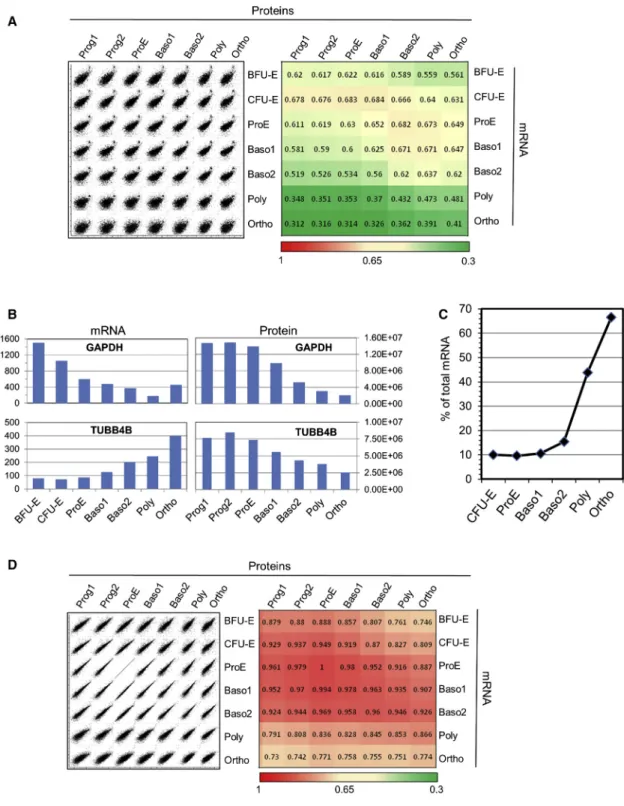
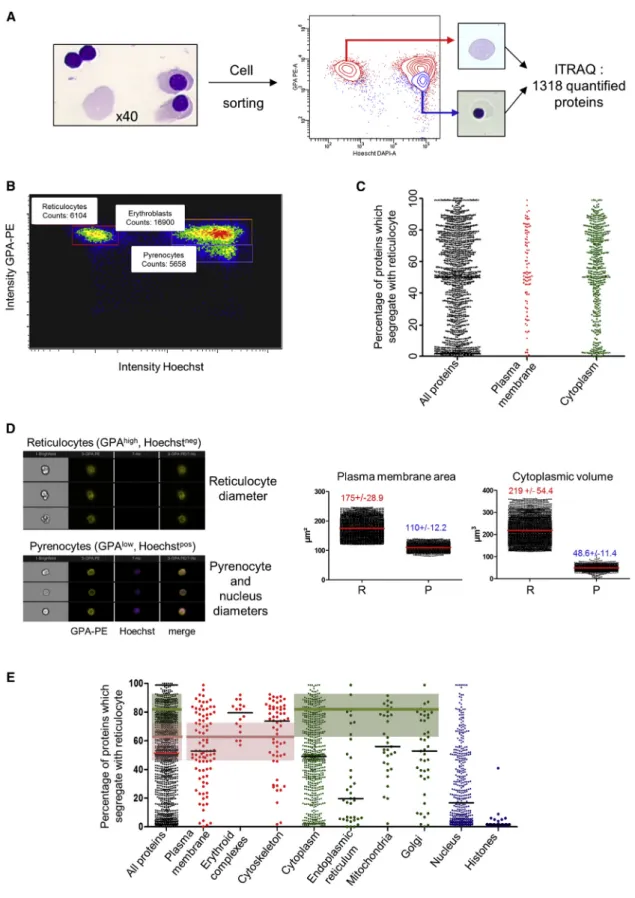
Figure
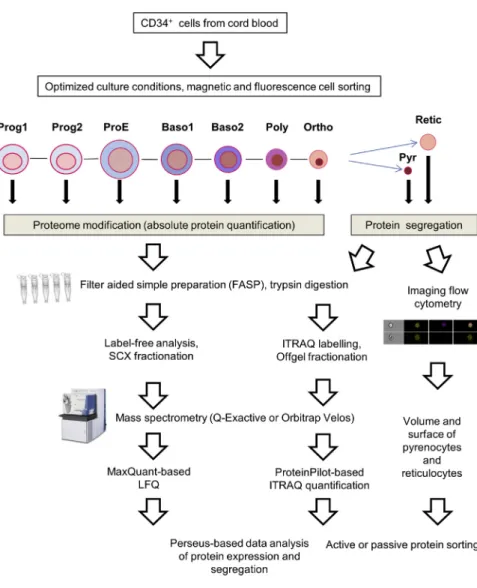
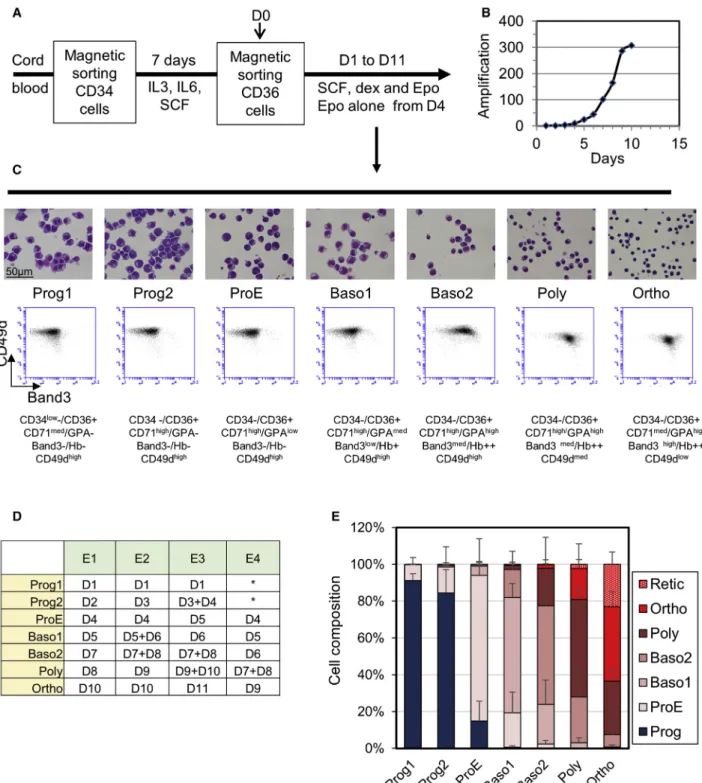
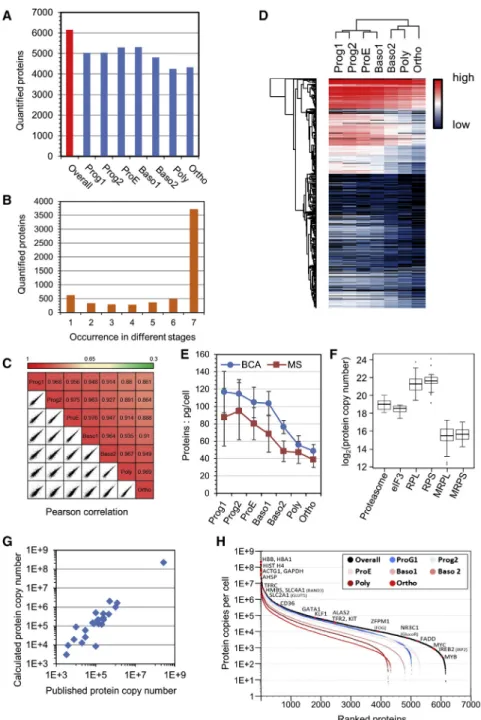
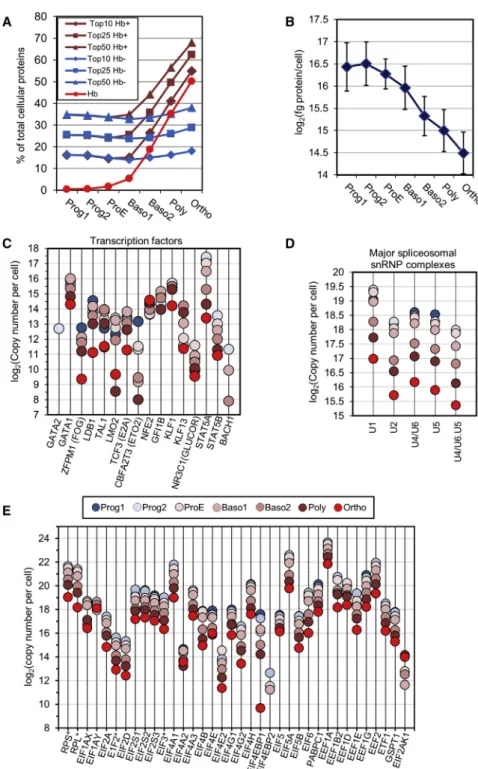
Documents relatifs
We introduce in this paper a technique in which we apply correlation analysis using only one execution power curve during an exponentiation to recover the whole secret
However, as horizontal CPA requires a single execution power trace to recover the secret exponent, DSA and Diffie-Hellman exponentiations are prone to this attack and other
(c) Each data matrix consisted of a 165 × 15 matrix of val- ues derived from fMRI responses of 10 subjects in response to 165 sounds.. Prior to MCCA the 6309 voxels were reduced to
Experimental analysis of masonry infilled frames using digital image
The usual approach is to build a 3-D reconstruction of the surface(s) from which all shape properties will then be derived without ever going back to the original images. In this
Conclusion: Human Gene Correlation Analysis (HGCA) is a tool to classify human genes according to their coexpression levels and to identify overrepresented annotation terms
In Figure 7A, the profiles of the dimensionless water vapor concentration in the lumen, during inspiration and expiration, are presented for three values of Re insp 1 /β
More particularly, we measure the correlation be- tween the ranking of low-, medium-, and high-quality limited-size approximation sets with respect to inverted generational
