HAL Id: tel-02164724
https://tel.archives-ouvertes.fr/tel-02164724
Submitted on 25 Jun 2019HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
squalene-adenosine nanoparticles : mechanism of action
and formulation
Marie Rouquette
To cite this version:
Marie Rouquette. Moving forward in the pre-clinical development of squalene-adenosine nanoparticles : mechanism of action and formulation. Galenic pharmacology. Université Paris Saclay (COmUE), 2019. English. �NNT : 2019SACLS035�. �tel-02164724�
Moving forward in the
pre-clinical development of
squalene-adenosine nanoparticles:
mechanism of action and
formulation
Thèse de doctorat de l'Université Paris Saclay préparée à la Faculté de Pharmacie de l’Université Paris-SudÉcole doctorale n°569 Innovation thérapeutique : du fondamental à l'appliqué (ITFA)
Spécialité de doctorat : Pharmacotechnie et biopharmacie
Thèse présentée et soutenue à Châtenay-Malabry, le 8 février 2019, par
Marie Rouquette
Composition du Jury : Philippe Barthélémy
Professeur, Université de Bordeaux (U1212) Rapporteur
Ling Peng
Directrice de recherche, CNRS (UMR7325) Rapporteur
Karine Andrieux
Professeur, Université Paris Descartes (UMR8258) Examinateur
Elias Fattal
Professeur, Université Paris-Sud (UMR8612) Président
Harivardhan Lakkireddy
Head of Formulation & Analytic Physicochemistry, Sanofi Examinateur
Fabienne Testard
Chercheuse, CEA (LIONS) Examinateur
Patrick Couvreur
Professeur, Université Paris-Sud (UMR8612) Directeur de thèse
Sinda Lepetre-Mouelhi
Maître de Conférences, Université Paris-Sud (UMR8612) Co-Directeur de thèse
NNT : 2 01 9 S A C L S 03 5
« La théorie, c'est quand on sait tout et que rien ne fonctionne. La pratique, c'est quand tout fonctionne et que personne ne sait pourquoi. Ici, nous avons réuni théorie et pratique : Rien ne fonctionne... et personne ne sait pourquoi ! »
Remerciements
Je tiens tout d’abord à remercier l’ensemble des membres de mon jury de thèse : le Pr.
Philippe Barthélémy et le Dr. Ling Peng pour me faire l’honneur d’évaluer mon travail en tant
que rapporteurs ; le Pr. Karine Andrieux, le Pr. Elias Fattal, le Dr Harivardhan Reddy et le
Dr Fabienne Testard pour avoir accepté de porter un regard critique sur cette thèse en tant
qu’examinateurs.
Je tiens également à remercier très chaleureusement Elias pour son accueil au sein du laboratoire. Tu ne m’as pas uniquement accueillie en tant que directeur de l’institut mais aussi en tant que simple collègue, et tu as discrètement veillé sur moi tout au long de ces trois ans (et peut-être encore plus au cours de ces derniers mois). Merci pour cette attention.
Un immense merci à Patrick, mon directeur de thèse, pour sa disponibilité au cours de ces trois ans. Votre emploi du temps est chargé mais vous avez une « disponibilité d’esprit » exceptionnelle : vous avez toujours été extrêmement réactif quand j’avais besoin de vous, et aviez toujours parfaitement à l’esprit l’état d’avancement de mes travaux. C’est une qualité exceptionnelle chez un responsable et je tenterai de la reproduire dans ma vie professionnelle future. Merci également pour toutes nos discussions, scientifiques ou non. J’aime écouter des histoires et vous savez en raconter de fameuses !
Je souhaite également témoigner toute ma gratitude envers Sinda, ma co-directrice de thèse. Tu cherches avant tout à soutenir et faire progresser les gens qui t’entourent, et tu le fais avec beaucoup d’humilité. Les personnes d’une telle générosité pour l’humain sont rares et précieuses. J’essaierai de garder en moi cet apprentissage, car je suis convaincue qu’une de nos missions professionnelles les plus importantes (mais également des plus faciles à oublier) est de nous aider les uns les autres à grandir.
Merci à Karine, Mélanie et toute leur équipe pour leur accueil particulièrement chaleureux à la plateforme animalerie de l’IGR. Malgré toutes les galères qu’on a traversées, ça a toujours été un bonheur de venir chez vous car je savais que j’y retrouverais votre bonne humeur inaltérable. Merci de m’avoir tant appris sur la souris et de m’avoir fait confiance dès le début pour la chirurgie.
I would also like to thank Pr. Adriaan P. IJzerman for welcoming me in his lab in Leiden for two weeks and taking time to discuss with me. Xue, you have been a wonderful teacher and host, and this collaboration could not have been as successful without you. I also would like to thank Rongfang for making preliminary experiments before I came, Henk de Vries, the one who knows everything about the lab, and Anna for giving me some tips on adenosine agonists and antagonists.
Je n’aurais pu obtenir tous ces résultats sans l’aide de mes stagiaires, master 2 ou BTS. Ils m’ont beaucoup appris et m’ont permis de faire mes premiers pas en tant qu’encadrante sans être trop sévères avec moi. Merci Manon pour tout ce boulot en culture cell, et Ophélie qui est maintenant bien plus calée en HPLC que moi ! Merci d’avoir gardé votre bonne humeur et votre confiance en ma capacité à vous guider malgré les moments difficiles. Une petite pensée également pour Yannis, mon stagiaire de troisième, qui m’a rappelé à quel point je trouve la biologie extraordinaire.
J’ai été entourée de personnes remarquables au sein de cet institut : Simona, la mémoire vivante de tout ce qui a été fait sur le squalène depuis la nuit des temps (et surtout depuis que le labo a développé la squalénisation), toujours prête à prendre du temps pour écouter les difficultés et aider ; Juliette, la pro du Western (en tournant ça comme ça on pourrait t’imaginer avec deux colts à la ceinture et un chapeau de cowboy, mais ça ne serait pas une image très fidèle à ta gentillesse !) ; Stéphanie, avec qui on a vécu les coupures d’électricité à répétition (et on n’a même pas fait de burn out !), merci pour les petits dessins sur les flasques ; Julie, the HPLC expert comme dirait Ophélie, et aussi celle qui sait à chaque instant où est Sinda ; Claudie, à l’écoute si attentive ; Didier, qui déjeune tous les jours au CROUS ; Assia et Ghozlene, grâce à qui j’ai pu faire toutes mes manips en radioactivité (et ce jusqu’au bout !) ; Jean-Philippe, dont la patience n’a d’égale que sa dextérité à l’AFM.
Merci également aux responsables des plateformes de la faculté de pharmarcie, notamment Delphine, toujours prête à aider, même dans le bus, et Valérie Nicolas, aux explications limpides concernant la microscopie confocale.
Vient ensuite la foule des doctorants que j’ai côtoyés avec grand plaisir au cours de ces trois dernières années. Johanna, bien sûr, sans qui je ne me serais pas lancée dans ma grande carrière artistique à l’institut. Merci d’avoir proposé de m’aider pour le film des 30 ans, je n’y
qui aurait été extrêmement triste pour l’ensemble de nos fans. Marion, qui a la capacité extraordinaire de râler toute la journée avec motivation, entrain et bonne humeur. Je n’ai toujours pas compris comment tu faisais ! Gianpiero, qui a illuminé de son amitié mes longs trajets en bus entre Paris et la lointaine Châtenay ; Jiao, for kindly sharing some of your culture (and some of your office !) with me ; Elise, notre maman qu’on aurait bien gardée un an de plus ;
Daniele, qui me comprend maintenant même quand je n’articule pas ; Julie (eh oui c’est ton
côté un peu schizo, tu es à la fois avec les statutaires et les doctorants), ta persévérance va payer ;
Justin, j’ai une immense admiration pour toi, tous les efforts que tu fais pour apprendre le
français, ta curiosité, ton attention à l’autre… tu es l’une des personnes qui m’a le plus impressionnée ici et je ne te l’ai sûrement pas suffisamment dit ! ; Flavio, cher compagnon d’adénosine-squalène, bon courage pour la fin ; Romain, garde ta bonne humeur et ta simplicité, elles sont des atouts extraordinaires ; Fanny et Annaëlle, avec qui ça a été un plaisir de partager le bureau ; Alex, toujours de bonne humeur, toujours motivé, toujours attentif à l’autre ; Tanguy, mon pharmacien préféré ; Raul, mon herboriste préféré ; Quentin, mon véto préféré ; et Julien, que j’aurais bien croisé plus souvent. Je ne peux pas citer tous les
doctorants, post-doctorants et stagiaires de l’institut dont j’ai croisé la route, pourtant vous
avez chacun contribué à créer cette ambiance si joyeuse et dynamique au sein du labo. Merci d’avoir fait de mon environnement de travail un environnement affectif.
J’ai également une pensée amicale pour l’équipe iGEM Paris Saclay 2016, que j’ai suivie dans la grande aventure iGEM lors de ma première année de thèse. J’ai adoré participer à toutes ces réunions avec vous, à ce mois de manips en juillet (où j’ai probablement appris autant que vous sur la biologie synthétique !), vous coacher pour la présentation finale à Boston, bref vous apporter mon maigre soutien pour ce projet fabuleux ! Vous suivre a été l’une des expériences qui m’a le plus appris en termes d’encadrement au cours de cette thèse, et je vous en suis très reconnaissante.
Je remercie ma famille et mes amis, qui m’ont entourée tout au long de ces trois ans, qui m’apportent chaque jour un nouvel entrain, de nouveaux projets, et qui m’aident à grandir tout au long de ma vie.
Enfin, je ne remercierai jamais assez mon mari, Bertrand, pour m’apporter le bonheur extraordinaire de partager sa vie avec moi. Au cours de ces trois ans de thèse, tu m’as offert par deux fois la beauté d’un engagement à vie l’un avec l’autre, en m’épousant et en me donnant un enfant.
Et merci finalement à cet enfant pour la confiance que tu m’accordes en grandissant jour après jour en moi. La mission que tu me confies, celle d’être mère, est probablement l’une des plus belles qui me sera donnée de vivre.
Table des matières
Table des matières ... 1
Abréviations... 5
Introduction générale ... 9
Introduction bibliographique ... 13
Adenosine and lipids: a forced marriage or a love match? ... 14
Abstract ... 14
I. Adenosine and adenosine derivates loaded liposomes ... 17
1) Liposomal adenosine ... 20
2) Liposomal ADP ... 21
3) Liposomal ATP... 22
II. Adenosine-lipid conjugates or nucleolipids ... 26
1) General synthetic strategies for adenosine nucleolipids ... 26
2) Adenosine nucleolipids self-assembly ... 27
3) Adenosine nucleolipids as delivery systems ... 33
III. Conclusion ... 37
IV. References ... 38
Chapitre 1... 47
Squalene-adenosine nanoparticles: ligands of adenosine receptors
or adenosine prodrug? ... 48
Abstract ... 48
I. Introduction ... 49
II. Materials and methods ... 50
1) Materials ... 50
2) SQAd NPs preparation ... 51
3) Radioligand displacement assay ... 52
4) Cell culture ... 52
5) Cell uptake of radiolabeled SQAd NPs and free adenosine ... 53
6) Cell uptake of fluorescently labeled SQAd NPs ... 53
8) Adenosine extracellular release ... 54
9) HPLC conditions ... 55
10) Intracellular cAMP levels ... 55
III. Results ... 55
1) SQAd NPs ... 55
2) SQAd interaction with human adenosine receptors ... 56
3) SQAd NPs internalization ... 57
4) SQAd degradation in acidic conditions ... 60
5) Adenosine extracellular release ... 60
6) Adenosine receptors activation ... 62
IV. Discussion ... 63
... 65
Acknowledgements ... 65
Authors contributions... 66
V. References ... 66
VI. Supplementary materials ... 68
Chapitre 2... 71
Towards a clinical application of freeze-dried squalene-based
nanomedicines ... 72
Abstract ... 72
I. Introduction ... 73
II. Materials and methods ... 74
1) Materials ... 74
2) SQAd NPs preparation ... 75
3) Dynamic light scattering (DLS) ... 75
4) Freeze-thawing and freeze-drying experiments ... 76
5) Quantitative determination of residual ethanol ... 76
6) Quantitative determination of residual moisture ... 76
7) Small-angle neutron scattering (SANS) ... 76
8) Small-angle X-ray scattering (SAXS) ... 77
9) Atomic force microscopy (AFM) ... 78
10) Cytotoxicity study... 78
11) Hepatotoxicity study ... 78
III. Results ... 80
2) Characterization of freeze-dried NPs after redispersion ... 81
3) NPs size characterization by DLS ... 81
4) NPs size characterization by small-angle neutron scattering (SANS) ... 82
5) NPs morphology after freeze-drying ... 83
6) Influence of residual ethanol on NPs size... 84
7) Influence of residual ethanol and freeze-drying on NPs supramolecular structure . 85 8) Long-term conservation ... 86 9) NPs innocuity ... 87 IV. Discussion ... 89 V. Conclusion ... 92 Acknowledgements ... 93 VI. References ... 93
VII. Supplementary materials ... 96
Discussion Générale... 97
I. SQAd NPs for the treatment of liver ischemia ... 98
1) SQAd NPs and liver ischemia ... 98
2) Ethanol toxicity ... 100
3) Freeze-drying as a way of removing ethanol ... 101
II. Understanding SQAd NPs mechanism of action ... 102
1) SQAd NPs vs. free adenosine vs. adenosine receptor agonists and antagonists ... 103
2) Adenosine release from SQAd NPs under ischemia ... 104
3) Liver is not the brain ... 105
III. Conclusion and perspectives ... 106
IV. References ... 108
Abréviations
[3H]DPCPX 1,3-[3H]-dipropyl-8-cyclopentylxanthine [3H]PSB11 [3 H]8-Ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]-purin-5-one [3H]PSB603 [3 H]8-(4-(4-(4-Chlorophenyl)piperazide-1-sulfonyl)phenyl)-1-propylxanthine [3H]ZM241385 [2-3 H]-4-(2-[7-amino-2-{2-furyl}{1,2,4}triazolo{2,3-a}{1,3,5,}triazin-5-yl amino]ethH]-4-(2-[7-amino-2-{2-furyl}{1,2,4}triazolo{2,3-a}{1,3,5,}triazin-5-yl)phenol Ad Adenosine AC Adenylyl cyclaseADA Adenosine deaminase
ADAC Adenosine amine congener
ADP Adenosine diphosphate
AMP Adenosine monophosphate
AMPC12 Adenosine 5'-dodecylphosphate
AMPC16 Adenosine 5'-hexadecylphosphate
AMPC18 Adenosine 5'-octadecylphosphate
AR Adenosine receptor
ATP Adenosine triphosphate
BCA Bicinchoninic acid
bFGF Basic fibroblast growth factor
cAMP Cyclic AMP
CHEMS Cholesteryl hemisuccinate
CHO Chinese hamster ovary cells
Chol Cholesterol CholEsteryl BODIPY™ FL C12 CholEsteryl 4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-dodecanoate CPA N6-cyclopentyladenosine
DAPI 4′,6-diamidino-2-phenylindole DDPA 1,2-didecanoyl-sn-glycero-3-phosphatidyl-adenosine DDPC 1,2-didecanoyl-sn-glycero-3-phosphocholine DHSG 1,5‐dihexadecyl‐N‐succinyl‐l‐glutamate diC8PA 1,2-dioctanoyl-sn-glycero-3-phosphatidyl-adenosine diC8PC 1,2-dioctanoyl-sn-glycero-3-phosphocholine diC8PU 1,2-dioctanoyl-sn-glycero-3-phosphatidyluridine
DLS Dynamic light scattering
DLPA l,2-dilauroyl-sn-glycero-3-phosphatidyl-adenosine DLPC l,2-dilauroyl-sn-glycero-3-phosphocholine
DLPU l,2-dilauroyl-sn-glycero-3-phosphatidyl-uridine DMPA 1,2-dimyristoyl-sn-glycero-3-phosphatidyl-adenosine DMPC l,2-dimyristoyl-sn-glycero-3-phosphocholine
DMSO Dimethyl sulfoxide
DNA Deoxyribonucleic acid
DOAPC Di-oleyl-adenosine-phosphocholine
DODAB Dimethyldioctadecyl ammonium bromide
DOP-Ade 1,2-dioleoyl-sn-glycero-3-phosphatidyl-adenosine DOPE 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine DOPC 1,2-dioleoyl-sn-glycero-3-phosphocholine DOPC-E 1,2-dioleoyl-sn-glycero-3-ethylphosphocholine DOTAP 1,2-dioleoyl-3-trimethyl-ammonium-propane DPPA l,2-dipalmitoyl-sn-glycero-3-phosphatidyl-adenosine DPPC l,2-dipalmitoyl-sn-glycero-3-phosphocholine DSPC 1,2-distearoyl-sn-glycero-3-phosphocholine DSPE-PEG 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-n-[methoxy(polyethylene glycol)] DSPG 1,2-distearoyl-sn-glycero-3-phosphorylglycerol
EBM-2 Endothelial basal medium
EHNA Erythro-9-(2-hydroxy-3-nonyl)-adenine
ENT Equilibrative nucleoside transporter
FAAL Fatty acyl-AMP ligase
FACS Fatty acyl-CoA synthetases
FBS Fetal bovine serum
GP Glycoprotein
GPCR G protein-coupled receptor
GUV Giant unilamellar vesicle
H12-PEG-Glu2C18 Dodecapeptide HHLGGAKQAGDV-polyethylene glycol-Glutamate 2C18
HPLC High-performance liquid chromatography
HSPC Hydrogenated soy phosphatidyl choline
IBMX Isobutyl-1-methylxanthine
Ki Ligand binding affinity
LDL Low-density lipoprotein
LDLR Low-density lipoproteins receptor
LPDS Lipoprotein deficient serum
NECA 5’-N-ethylcarboxamidoadenosine NP Nanoparticle NSB Non-specific binding ODA Octadecylamine PAMAM Poly(amidoamine) PAPC 2′,3′-O-16-hentriacontanyliden-adenosine-5′-phosphocholine
PBS Dulbecco’s phosphate buffered saline
PC Phosphatidylcholine
PdI Polydispersity index
PEG-PE Polyethylene glycol-phosphatidylethanolamine
pKi - Log(Ki)
polyU Polyuridylic acid
POPA 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidyl-adenosine POPC 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine POPG 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidyl-glycerol POPU 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidyl-uridine PS Phosphatidylserine PUPC 2′,3′-O-16-hentriacontanyliden-uridine-5′-phosphocholine
RLU Relative Luminescence Unit
RNA Ribonucleic acid
Ro 20-1724 4-(3-butoxy-4-methoxybenzyl) imidazolidone SAPC 2’,3’-O-18-pentatriacontanyliden-adenosine-5’-phosphocholine SM4 Sulfatide SQAd Squalene-adenosine SUPC 2’,3’-O-18-pentatriacontanyliden-uridine-5’-phosphocholine TB Total binding
WGA Wheat germ agglutinin
XAC Xanthine amine congener
ZM241385 4-(-2-[7-amino-2-{2-furyl}{1,2,4}triazolo{2,3-a}{1,3,5}triazin-5-yl-amino]ethyl)phenol
Introduction générale
Le développement de nouvelles techniques de délivrance du médicament permet d’adapter la stratégie d’administration des substances actives en fonction de leurs propriétés physico-chimiques et de la cible thérapeutique envisagée. En particulier, l’introduction des nanomédecines (nanotechnologies appliquées au domaine de la médecine) dans les années 60 a été la source d’importants espoirs pour le traitement de maladies graves telles que le cancer. Cependant, bien qu’une quinzaine de ces nanoformulations aient été mises sur le marché, le développement des nanomédecines en général a rencontré plusieurs verrous technologiques. En effet, les nanomédicaments présentent souvent un très faible taux de charge en substance active, relarguent parfois de manière massive et précoce le principe actif (phénomène de « burst release ») et peuvent exhiber une certaine toxicité due au matériel de vectorisation utilisé. De plus, la transposition d’échelle pour une production industrielle se révèle parfois trop compliquée et/ou coûteuse.
La stratégie de « squalénisation », mise au point depuis 2004 par l’équipe du Professeur Couvreur, a permis de lever certains de ces verrous. En effet, cette technique consiste à coupler une substance active à un dérivé du squalène – un lipide endogène, biocompatible et biodégradable, précurseur du cholestérol – afin de former un bioconjugué ayant la capacité de s’auto-assembler en nanoparticules d’une centaine de nanomètres de diamètre en milieu aqueux. Ces nanomédicaments présentent notamment un fort taux de charge en principe actif, une très bonne biocompatibilité, une pharmacocinétique nettement améliorée par rapport à la substance active libre et une biodistribution beaucoup plus ciblée de celle-ci.
L’application de cette stratégie à l’adénosine, une molécule au potentiel thérapeutique important mais limité par un temps de demi-vie plasmatique extrêmement court, a fourni des résultats particulièrement prometteurs dans le cadre du traitement de l’ischémie cérébrale et du traumatisme de la moëlle épinière. L’objectif de cette thèse était donc de faire progresser le développement pré-clinique de ces nanoparticules d’adénosine-squalène selon deux axes majeurs : l’étude de leur mécanisme d’action et l’optimisation de leur formulation.
Dans une première partie bibliographique, nous présenterons les différentes associations créées jusqu’à aujourd’hui entre l’adénosine et les lipides. Nous aborderons notamment la délivrance de l’adénosine et de ses dérivés phosphorylés via des liposomes, puis nous décrirons les différents bioconjugués lipidiques formés à base d’adénosine ainsi que leurs capacités d’auto-organisation supramoléculaire.
Le premier chapitre expérimental décrira ensuite les résultats obtenus lors de l’étude in
vitro du mécanisme d’action des nanoparticules d’adénosine-squalène. L’interaction directe
entre l’adénosine-squalène (sous forme de nanoparticule ou de bioconjugué) et les récepteurs à l’adénosine a été étudiée grâce à une expérience de déplacement d’un radioligand. Cette interaction s’étant révélée négligeable, l’étude de l’adénosine-squalène en tant que prodrogue a été réalisée sur des hépatocytes. L’adénosine-squalène est en effet retrouvé en quantités importantes dans le foie après injection des nanoparticules par voie intraveineuse chez la souris. Ainsi, l’internalisation par les cellules HepG2 de nanoparticules d’adénosine-squalène radiomarquées ou fluorescentes, suivie du relargage d’adénosine, ont été étudiés. Cette étude a permis de mettre en lumière la formation d’un réservoir intracellulaire d’adénosine due à l’internalisation puis à l’accumulation des nanoparticules d’adénosine-squalène.
Le second chapitre expérimental présentera enfin l’optimisation des conditions de lyophilisation des nanoparticules d’adénosine-squalène. La bonne conservation de leurs propriétés physico-chimiques a été vérifiée par différentes techniques analytiques, dont la diffusion dynamique de la lumière, la diffraction des rayons X et des neutrons aux petits angles, ou encore la microscopie à force atomique. L’influence de la présence d’éthanol résiduel (utilisé lors de la formulation) sur le résultat de la lyophilisation est un point intéressant qui a été soulevé lors de cette étude. La lyophilisation a permis une bonne stabilité des nanoparticules dans le temps avec une reconstitution très aisée de celles-ci, les rendant ainsi très facilement manipulables en vue d’une administration en cas d’urgence.
L’ensemble de ces travaux constitue ainsi une avancée significative dans le développement pré-clinique des nanoparticules d’adénosine-squalène, par la mise en lumière de leur mécanisme d’action et l’établissement d’une nouvelle formulation, plus pratique et reproductible.
General Introduction
Ongoing advances in drug delivery help to adapt the administration strategy of the active ingredient taking into account its physico-chemical properties as well as the therapeutic target. In particular, the introduction of nanomedicines (or nanotechnologies applied to medicine) in the 1960’s has brought hope for enhancing the effectiveness of the treatments against severe diseases such as cancer, through a better targeted approach. However, even though around fifteen nanoformulations are already used in the clinic, the development of nanomedicines in general has suffered from serious drawbacks. Indeed, nanomedicines often present extremely low drug loadings, may prematurely and massively release the active compound before reaching the target (phenomenon of “burst release”) and exhibit some toxicity due, among other things, to the vector per se. Moreover, scaling-up the production for clinical needs is sometimes challenging and/or too costly.
The so-called « squalenoylation » strategy, developed since 2004 by Pr. Couvreur’s team, has overcome some of these limitations. This original technology consists in coupling an active ingredient with a derivative of squalene – an endogenous, biocompatible and biodegradable lipid which is a precursor of cholesterol – to form bioconjugates able to self-assemble as nanoparticles of around 100 nm of diameter in aqueous medium. The resulting nanomedicines generally present high drug loadings, very good biocompatibility, enhanced pharmacokinetics compared to the free drug as well as a more targeted biodistribution.
This strategy was applied to adenosine, a molecule which high therapeutic potential is severely impaired by its very short half-life in blood circulation. Squalene-adenosine nanoparticles have shown extremely promising results for the treatment of cerebral ischemia and spinal cord injury. Hence, the objective of this thesis was to further develop squalene-adenosine nanoparticles at the pre-clinical stage, pursuing two major goals: studying their mechanism of action and optimizing their formulation.
First, in a bibliographic part, we will present the different associations which have so far been designed between adenosine and lipids. More precisely, we will address liposomal
delivery of adenosine and its phosphorylated derivatives, before describing adenosine-based lipid bioconjugates and their supramolecular self-assemblies.
The first experimental chapter will then describe the mechanism of action of adenosine nanoparticles through in vitro studies. Direct interaction between squalene-adenosine (whether as a nanoparticle or bioconjugate) with squalene-adenosine receptors was studied by radioligand displacement assays. As this interaction was found to be negligible, we therefore investigated whether squalene-adenosine could act as a prodrug. This mechanism was studied on hepatocytes, as high quantities of squalene-adenosine are found in the liver after intravenous injection of squalene-adenosine nanoparticles in mice. HepG2 cellular uptake of radio- or fluorescently labeled squalene-adenosine nanoparticles was studied, as well as the resulting adenosine release. This study highlighted the presence of an adenosine intracellular reservoir due to the internalization and intracellular accumulation of squalene-adenosine nanoparticles.
Finally, the second experimental chapter will present the optimization of squalene-adenosine nanoparticles formulation through the use of the best freeze-drying conditions. The preservation of the nanoparticles’ physico-chemical properties was checked by different analytical techniques, such as dynamic light scattering, small-angle X-ray or neutrons scattering or atomic force microscopy. In addition, the presence of residual ethanol (used during the nanoparticles’ formulation) was shown to influence the lyophilization success. Of note, this finding had rarely been studied before. In that respect, the freeze-drying of nanoparticles led to their long-term stability as well as to their easy and rapid reconstitution in ready-to-use nanoparticulate suspension which could thus prove quite convenient for administration in emergency cases.
Overall, this work represents a major step forward the pre-clinical development of squalene-adenosine nanoparticles by bringing to light their mechanism of action and establishing a new, easy-to-use and reproducible formulation.
Introduction bibliographique
Adenosine and lipids: a forced marriage
or a love match?
Marie Rouquette, Sinda Lepetre-Mouelhi, Patrick Couvreur
Institut Galien Paris-Sud, Université Paris-Sud, CNRS, Université Paris Saclay, Châtenay-Malabry, France.
Adenosine and lipids: a forced marriage
or a love match?
Abstract
Adenosine is a fascinating compound, crucial in many biochemical processes: this ubiquitous nucleoside serves as an essential brick for building RNA, is also a component of ATP and regulates numerous pathophysiological mechanisms via binding to four extracellular receptors. Due to its hydrophilic nature, it belongs to a different world than where lipids come from, and has no affinity for them. Since the 1970’s, however, new discoveries have emerged and prompted the scientists to associate adenosine with the lipid family, especially via liposomal preparations and bioconjugation. This seems to be an arranged marriage, but could it turn into a true love match? This review clustered all types of unions which were established between adenosine and lipids. Even though exciting supramolecular structure were observed with adenosine-lipid conjugates, as well as, with liposomal preparations which resulted in promising pre-clinical results, the translation of these technologies to the clinic is still limited.
Adenosine is a nucleoside involved in multiple essential biochemical processes both intra- and extracellularly (Figure 1). In the cell, adenosine can be phosphorylated into adenosine monophosphate (AMP), which is used by the cell as an RNA building block. This phosphorylation process can be repeated in order to generate adenosine triphosphate (ATP), the “molecular currency” furnishing energy to the cell. ATP can also be converted by adenylyl cyclase (AC) into cyclic AMP (cAMP), a very common second messenger which triggers intracellular signal transduction cascade. Outside the cell, adenosine regulates essential processes by binding to four G-protein-coupled receptors: A1, A2A, A2B and A3 [1], which govern
major pathophysiological functions. Equilibrative nucleoside transporters (ENTs), which are widely expressed by the cells [2], maintain the balance between intra- and extracellular
concentrations dramatically rise due to the release of abundant intracellular ATP by damaged cells [3] and its prompt transformation into adenosine by ectonucleotidases CD39 and CD73.
Adenosine is a hydrophilic molecule that has no affinity with lipids. Like Romeo and Juliet, they come from two separate worlds that rarely mingle. But, if forced to join, would they repel each other, accommodate or start a fusional relationship? Scientific discoveries in the 70’s and 80’s encouraged research on that topic.
The development of liposomes for drug delivery in the early 1970’s [4] was seen by some specialists as a very suitable match for adenosine and its derivatives. Indeed, adenosine and ATP have very short plasmatic half-life [5,6] due to their degradation by plasmatic enzymes and rapid uptake by erythrocytes and endothelial cells, which could be circumvented by liposomal encapsulation. First proposed applications were the delivery of liposomal ATP to treat rat ischemic brains [7] and liposomal adenosine to examine its effect on the contraction of rat
Figure 1 Intracellular and extracellular adenosine. In the cell, adenosine is degraded by adenosine
deaminase (ADA) into inosine or phosphorylated by adenosine kinase (AdK) into adenosine mono-phosphate (AMP), di-mono-phosphate (ADP) or tri-mono-phosphate (ATP), which is used as substrate by adenylyl cyclase (AC) for cyclic AMP (cAMP) formation. Outside the cell, adenosine binds four receptors involved in various pathophysiological processes.
aortic smooth muscle cells [8]. As will be presented in the first part of this review, the first matchmakers’ intuition was accurate and liposomal forms of adenosine, ATP and even ADP have been developed and studied since this consensual marriage with expected successful outcomes.
Completely independently, a few naturally occurring nucleolipids were discovered in the 1970’s, such as tunicamycins, which were isolated from Streptomyces lysosuperficus [9]. This drove chemists to create multiple artificial nucleolipids a few years later, including adenosine nucleolipids like adenosine-5’-alkylphosphates [10]. There were two main rationales for studying such adenosine nucleolipids: (i) authors saw them as possible intermediates in enzymatic reactions involving ATP and lipids and wanted to study their physiological significance [10,11], or (ii) they wanted to explore the existence of hydrophobic regions in the active sites of ATP-dependent enzymes [12].
With a totally different mindset, a third rationale for developing adenosine nucleolipids was established in the late 1980’s. Adenosine congeners were functionalized by covalently attaching lipids at a permissive site (e.g. N6 or C2 positions of adenosine) with the idea to alter the drug distribution and metabolism in vivo while conserving good binding properties without any prior cleavage [13,14].
Even though these approaches have opened the field of adenosine nucleolipids and given some interesting results, no author really anticipated that this forced marriage would become a perfect love match between adenosine and lipids. Nucleolipids, and adenosine nucleolipids in particular, present remarkable properties that will be exposed in the second part of this review.
I. Adenosine and adenosine derivates loaded liposomes
Liposomes are lipid-bilayer vesicles made of combinations of natural or/and synthetic lipids. These lipids are perfectly biocompatible and biodegradable and have already been used as excipients in numerous vaccines. As a result, liposomes can be used as safe delivery systems for both hydrophilic and hydrophobic molecules. Up until now, more than 15 liposomal formulations already reached the market, Doxil® being the most well-known among them [15].When adenosine, ADP or ATP are loaded into the liposomes’ hydrophilic core, they are protected from the blood stream and their pharmacokinetics and biodistributions are completely modified, offering new therapeutic possibilities. Table 1 describes the composition and size of the liposomes that will be described in this part, as well as their drug loading and/or encapsulation efficiency when available. Regrettably, not all reports provide a full and comprehensive characterization of their liposomal system, which leads to uncertainties in terms of dosage, especially because liposomal formulation often present very poor drug loadings.
Table 1 Adenosine, ADP and ATP-loaded liposomes.
Drug loaded Lipid composition size (nm) Liposome
Drug loading (µmol drug / µmol lipid)
Encapsulation
efficiency (%) target Therapeutic References
Adenosine
DSPC:DSPG:chol 168 ± 18 n.d. 12.9
Inflammation [16]
DSPC:chol 206 ± 40 n.d. 8.6
DSPC:DODAB:Chol 202 ± 27 n.d. 10.5
HSPC:chol:DSPE-PEG 134 ± 21 n.d. n.d. ischemia Myocardial [17]
Soybean oil:PC:chol:glycerin n.d. n.d. n.d. Osteoarthritis [18]
ADP DMPC:chol:DHSG:DSPE-PEG:H12-PEG-Glu2C18 240 ± 68 n.d. n.d. Hemostasis [19–22] DPPC:chol:DHSG:DSPE-PEG:H12-PEG-Glu2C18 285 ± 78 n.d. n.d. DSPC:chol:DHSG:DSPE-PEG:H12-PEG-Glu2C18 298 ± 107 n.d. n.d. ATP PC:chol:SM4 ~ 140 ≤ 0.39 n.d. Cerebral ischemia [7,23] PC:chol n.d. n.d. n.d. [24] PS ~ 100 n.d. n.d. Retinal ischemia [25,26] PC n.d. n.d. PS:PC n.d. n.d. PC:chol:stearylamine > 2000 0.32 ± 0.02 n.d. Myocardial ischemia [27] PC:chol:DSPE-PEG:DOTAP ~ 200 c.a. 0.40 n.d. [28,29] PC:chol:DSPE-PEG:2G4-PEG–PE ~ 200 n.d. n.d. [30] PC:chol:DSPE-PEG:DOTAP:2G4-PEG–PE 190 ± 45 c.a. 0.40 n.d. [31] PC:chol n.d. n.d. 8 Liver ischemia [32] PC:chol:PG ~ 200 0.35 ± 0.06 n.d. [33,34] PC:chol ~ 100 n.d. n.d. [35,36] PC:chol:PG n.d. n.d. PC:chol:ODA ≤ 0.07 ± 0.01 n.d.
PC:chol:DOTAP:DOPE n.d. n.d.
PC:chol:DOTAP:Lac-10-chol 0.04 ± 0.01 n.d.
PC:chol:DOTAP:DOPE:Lac-10-chol 0.03 ± 0.01 n.d.
DPPC:chol:PEG ~ 120 0.33 ± 0.11 n.d. transplantation Islet [37]
DSPC:DSPE-PEG:chol n.d. n.d. n.d. ischemia Intestinal [38]
VitaSol n.d. n.d. n.d. Wound healing [39] ATPv n.d. n.d. n.d. [40] DOPC:DOPC-E n.d. n.d. n.d. [41] PC:DOTAP 120 - 160 n.d. n.d. [42–45] ATP + pentobarbital + suramin
DOPE:CHEMS:PEG-PE ~ 150 n.d. n.d. ischemia Cerebral [46]
CHEMS: cholesteryl hemisuccinate; Chol: cholesterol; DHSG: 1,5-dihexadecyl-N-succinyl-l-glutamate; DODAB: dimethyldioctadecyl ammonium bromide; DOPE: 3-phosphoethanolamine; DOPC: 3-phosphocholine; DOPC-E: dioleoyl-sn-glycero-3-ethylphosphocholine; DOTAP: dioleoyl-3-trimethyl-ammonium-propane; DPPC: dipalmitoyl-sn-glycero-3-phosphatidylcholine; DSPC: distearoyl-sn-glycero-3-phosphocholine; DSPE-PEG: distearoyl-sn-glycero-3-phosphoethanolamine-n-[methoxy(polyethylene glycol)]; DSPG: 1,2-distearoyl-sn-glycero-3-phosphorylglycerol; H12-PEG-Glu2C18: dodecapeptide HHLGGAKQAGDV-polyethylene glycol-Glutamate 2C18; HSPC: hydrogenated soy phosphatidyl choline; n.d.: not determined; ODA: octadecylamine; PC: phosphatidylcholine; PEG-PE: polyethylene glycol-phosphatidylethanolamine; PG: phosphatidylglycerol; PS: phosphatidylserine; SM4: sulfatide.
1) Liposomal adenosine
The main objective of adenosine encapsulation into liposomes is to prolong its half-life in the blood stream. Liposomal formulations can also enhance the quantity of active drug at the site of action by passive or active targeting.
Based on this background knowledge, Gutman et al. developed adenosine loaded liposomes to target inflammation [16]. They showed that negatively charged liposomes composed of 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), distearoyl-phosphatidylglycerol (DSPG) and cholesterol could be prepared by reverse phase evaporation method and loaded with adenosine (reaching a drug loading charge of 12.9 %), and that these monodisperse liposomes of around 170 nm of diameter were internalized by human monocytes and reduced inflammation in vitro. Regrettably, the only parameter assessed in vivo was the absence of adenosine loaded liposomes toxicity on monocytes, whereas their influence on inflammation was not monitored.
Another approach for passive targeting via liposomal formulation was developed by Takahama et al. for myocardial ischemia and reperfusion [17]. It is known that cellular permeability is enhanced in the zone of infarct due to the disruption of the vascular endothelial integrity. As a result, the authors decided to prepare PEGylated adenosine liposomes with prolonged circulation time for reaching higher concentration of adenosine in the target zone. For this purpose, neutral monodisperse liposomes were prepared by the hydration method, with hydrogenated soy phosphatidylcholine, cholesterol and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-n-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2000). The use of fluorescent liposomes showed that they indeed accumulated in the necrotic area, which correlated with a prolonged residence time of liposomal radiolabeled adenosine in the same zone. This led to a reduction of the infarct size which was not associated with usual adenosine side effects (hypotension and bradycardia), confirming the advantage of using liposomal delivery.
Very recently, Corciulo et al. developed the idea of using liposomal adenosine for treating osteoarthritis [18], based on the fact that extracellular adenosine downregulation was known to be implicated in the development of the disease, more specifically via its interaction with A2A receptor. Unexpectedly, they observed that injection of A2AR agonists did not protect
liposomes made of soybean oil, phosphatidylcholine, cholesterol and glycerin and injected them locally in the joint. Using this formulation of adenosine, they witnessed important prevention and protection effects against the disease, with reduced joint swelling, cartilage and joint protection, as well as lowered OARSI scores, and demonstrated that these effects depended on A=R activation. Even though these results constitute a promising pre-clinical proof-of-concept, it seems unfortunate that liposomal formulations were so poorly defined (basic information on their composition can only be found in the corresponding patent [47]) and scarcely characterized in this work, with no mention of physico-chemical parameters.
2) Liposomal ADP
An original liposomal ADP system has been developed for 10 years in Takeoka’s lab for platelet substitution. Pegylated liposomes were loaded with ADP in the intraliposomal aqueous core and functionalized with a fibrinogen γ-chain peptide. This peptide corresponded to the carboxyl terminal 12 amino acids of the fibrinogen γ-chain and is a ligand for glycoprotein IIb/IIIa receptors which are expressed on activated platelets at the site of injury. As a result, these “H12-(ADP)-liposomes” specifically accumulated within the sites of injuries after intravenous injection [22] and reduced the bleeding time in thrombocytopenic rats [21] and rabbits [20,48] by playing two different roles: (i) they enhanced platelet aggregation via cross-linking between activated platelets thanks to their functionalization with the fibrinogen γ-chain peptide and (ii) they enhanced platelet activation by releasing ADP, which acts through platelet surface receptors P2Y1, P2Y12 or P2X1. Interestingly, authors could modulate this ADP release by tuning the vesicles lamellarity and flexibility [21].
Recently, this team made a brilliant conceptual and experimental move: instead of continuing to consider ADP only as a drug for platelet activation, they started looking at it as an adenosine prodrug. This led them to evaluate the pharmacological efficacy of H12-(ADP)-liposomes on blast lung injury, where ADP would be useful in treating the hemorrhage, while adenosine would exert anti-inflammatory protective effects [48] (Figure 2). They obtained an important increase in mice survival after lung injury in the group treated with H12-(ADP)-liposomes, going with reduced tissue damage/hemorrhage and inflammation. Moreover, they proved the implication of adenosine in this phenomenon, as treatment with A2A and A2B
3) Liposomal ATP
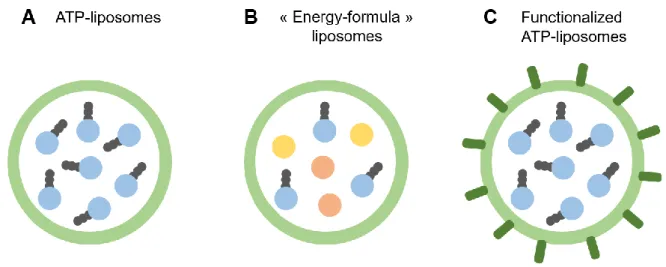
Like adenosine, ATP cannot be efficiently delivered intracellularly by itself. It is very rapidly metabolized in the bloodstream and the hydrophilic nature of this nucleoside prevents from crossing cell membranes. As a result, ATP-loaded liposomes have been used for several decades to improve ATP delivery (Figure 3A).
As ATP is the main energy currency in the cell, intracellular ATP delivery has mainly been used for pathologies involving a phenomenon of energy failure. The main application has been ischemia, i.e. the lack of blood supply in a tissue, leading to a shortage in oxygen, decreased cell metabolism and ATP depletion. Ischemic insults can virtually affect any organ or tissue, the most common and severe cases being heart, brain and liver ischemia.
Figure 2 Hypothetic mechanism of action of H12-(ADP)-liposomes. (1) H12-(ADP)-liposomes
accumulate at the site of injury by binding glycoprotein (GP) IIb/IIIa on activated platelets. (2) This also enhances platelet cross-linking, as the same liposome presents several fibrinogen γ-chain peptides moieties on its surface and can therefore bind several activated platelets. (3) ADP released from the liposomes (4) further activates platelets via P2Y1, P2Y12 or P2X1 receptors, and (5) is also metabolized into adenosine, which has anti-inflammatory protective effects.
Early proof of concept studies on ATP-liposomes pharmacological efficacy against cerebral ischemia were conducted on rats in the 1990’s. Liposomes efficiently protected ATP from plasma degradation, could cross the blood-brain-barrier under hypoxic conditions [24] and protected the rats against cerebral ischemic insults [7,23]. A closely related model of retinal ischemia [49] was later used to better understand the mechanism of action of these ATP-liposomes. ATP delivery led to increased cell survival and decreased cell necrosis in primary retinal ganglion submitted to glucose-oxygen deprivation in vitro, and to decreased neurons death in the ganglion cell layer in vivo after ischemia-reperfusion. Pro-inflammatory genes were also down-regulated in ischemic retinas following ATP-liposomes treatment [25,26]. These simple ATP-liposomes were recently redesigned into pH-sensitive “energy formula”-loaded liposomes [46] (Figure 3B). The lipid composition was chosen in such a way that liposomes responded to pH and released their content at the penumbral intracellular pH. In addition, ATP was co-loaded with pentobarbital, which inhibits energy consumption, and with suramin, for blocking ATP toxic extracellular actions. Even though these liposomes protected a neuronal cell line against ATP depletion in vitro, they failed to protect astrocytes or endothelial cells under the same conditions and no in vivo data were reported. Hence, the superiority of these “energy-formula”-liposomes, compared to simple ATP-liposomes, remains to be proven.
ATP-liposomes were also shown to accumulate in myocardial infarct tissues in dogs [27]. Torchilin’s team confirmed this result in isolated rat hearts perfused with a Langendorff instrument and showed that ATP-liposomes could protect the mechanical functions of the myocardium during ischemia/reperfusion [29]. Similarly, ATP-liposome treatment reduced the fraction of the irreversibly damaged heart area within the total area at risk in a model of myocardium infarction in rabbits [28]. Further improvement of the liposomes was realized by functionalizing them with the monoclonal 2D4 antibody which specifically binds cardiac myosin [30] (Figure 3C). Intracellular cardiac myosin become, indeed, exposed to the blood-stream after sarcolemmal disruption happening during ischemia. Hence, targeting these proteins should lead to higher accumulation of antibody-coated ATP-liposomes compared to bare ATP-liposomes. Even though this mechanism was not demonstrated in vivo, it was shown that the pharmacological efficacy of antibody-coated ATP-liposomes was enhanced compared to bare ATP-liposomes on isolated rat hearts perfused with a Langendorff instrument [31].
Regarding liver ischemia, ATP liposomes have shown protective activity on rat models of both warm ischemia in vivo after a hypovolemic shock [32] and cold ischemia ex vivo during liver preservation for transplantation [33,34]. In both cases, authors witnessed an enhanced preservation of the ATP cellular content. Functionalizing ATP-liposomes with an asialoglycoprotein receptor ligand did not show any improved hepatocytes targeting or delivery efficacy so far [35], and their freeze-drying led to ATP leakage [36].
Regarding organ preservation, protective effects of ATP-liposomes were also tested on a model of ischemic pancreatic β cells for possible application on islet transplantation [37]. ATP-liposomes were functionalized with a fibronectin-mimetic peptide, which favored their cell internalization. Surprisingly, metabolic protection against the ischemic insult was shown to arise from both ATP and the lipids constituting the liposome (l,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), cholesterol, PEG2000). However, ATP-liposomes could not restore insulin production from ischemic islet cells.
Finally, in a model of mesenteric ischemia/reperfusion, authors showed that liposomal ATP could restore crypt proliferation but did not prevent cell death in the ischemic gut, arguing towards important complementary cytoprotective roles of other glucose metabolites like pyruvate [38].
Figure 3 Different types of ATP-liposomes. (A) Basic ATP-loaded liposomes can be complexified by
(B) co-loading with complementary energy molecules like pentobarbital and suramin or (C) by functionalization with targeting moieties like antibodies or receptor ligands. The lipid bilayer composition can also be tuned to provide pH-sensitivity, fusogenicity or stealthiness.
In terms of commercial applications, highly fusogenic ATP-liposomes have been developed by the company Energy Delivery Solutions, a spin-off from Chien’s lab at the University of Louisville, under the product name of ATPv (formerly VitaSol). These liposomes are composed of a stable vesicle former, for example 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) or soy phosphatidylcholine (PC), and an unstable vesicle former, like 1,2-dioleoly-sn-glycero-3-ethylphosphocholine (DOPC-e) or 1,2-dioleoyl-3-trimethyl-ammonium-propane (DOTAP) [50]. Dressings were saturated with these ATPv and applied to wounds to study the liposomes effect on skin healing. Very promising results were obtained on wounds performed on mice heads [41] and on ischemic ears of normal [42,45] or diabetic [44] rabbits. A very special finding was the discovery of a completely new healing process, with extremely fast granulation attributed to massive macrophage migration, their in situ proliferation and direct collagen production [42–45]. On a different therapeutic area, using a model of hemorrhagic shock on rats, these ATP-liposomes also improved survival when injected during resuscitation [39,40]. ATPv liposomes are currently sold as holding solutions for hair transplants. Clinical trials on other applications are eagerly awaited.
In a completely different mindset, ATP-liposomes have also been used as a helper system for ATP-responsive nanocarriers. In this strategy, the delivery of an anticancer drug by fusogenic liposomes containing ATP-responsive DNA scaffold was enhanced by the co-administration of ATP-liposomes [51].
All these studies show that there are still interesting perspectives in ATP-liposomes and ATP-vesicles development, particularly by tuning the cargo composition with other energetic molecules, by decorating the carrier surface with specific peptides or antibodies for enhanced targeting, and by finding new areas of applications. A better understanding of the ATP-liposomes mechanism of action may help move this technology to the clinic. In particular, it would be interesting to confirm whether ATP-liposomes act only by delivering intracellular ATP or if this ATP delivery leads to adenosine extracellular delivery [52,53] as in the case of ADP-liposomes [48].
II.
Adenosine-lipid conjugates or nucleolipids
Nucleolipids are composite molecules formed by the assembly of a lipidic component and a nucleobase, nucleoside or nucleotide.
1) General synthetic strategies for adenosine nucleolipids
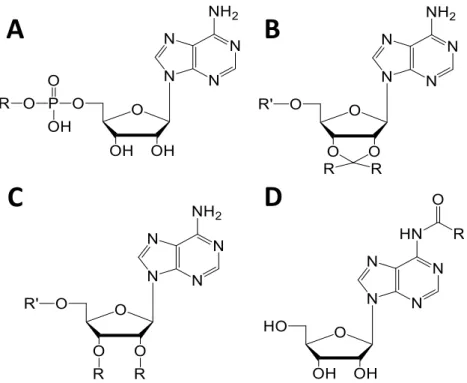
Different chemical approaches can be employed to obtain artificial adenosine nucleolipids. A commonly employed strategy was first proposed by Shuto and colleagues in 1987 [54] for obtaining 5’-phosphatidyl-nucleosides (Figure 4A). This method’s popularity arises from its simplicity: a single-step enzymatic reaction in a two-phase system (chloroform and buffer) is necessary, in which phospholipase D catalyzes the transfer reaction of the phosphatidyl residue to the primary hydroxyl group of nucleosides. One of the first artificial adenosine nucleolipid, l,2-dipalmitoyl-sn-glycero-3-phosphatidyl-adenosine (DPPA), was synthetized by this method with a yield of 52 %. A few years later, Itojima et al. made use of this technique for creating a library of four 5’-phosphatidyl-adenosines with different alkyl chain lengths [55].
Other organic strategies consist of activating either the adenosine monophosphate or the lipid moiety before coupling them. For example, 5’-phosphatidyl-adenosines can also be obtained by condensation of fatty alcohols with AMP activated by N,N'-dicyclohexylcarbodiimide [10,56] or by reaction of AMP tetrabutylammonium salt with alkyl bromides [57]. Ahlers et al. created a new type of adenosine nucleolipids (Figure 4B, R’=H) by coupling N-hydroxysuccinimide actived esters of 2',3'-O-(3-carboxy-l-methylpropyliden)adenosine [58] with mono or dialkylamines. More recently, Moreau et al. synthesized other types of adenosine nucleolipids (Figure 4B, R’=H) by reacting adenosine with dimethoxy ketal of palmitone [59]. These adenosine nucleolipids were also reacted with 2-chloro-1,3,2-dioxaphospholane-2-oxide and trimethylamine in order to obtain lipid adenosine phosphocholines (Figure 4B, R’=phosphocholine) [59]. The same team also used imidazole activated fatty acids with 2’,3’-(isopropylidene)-5’-(phosphocholine)-adenosine to form lipid adenosine phosphocholines (Figure 4C, R’=phosphocholine) [60]. Over the same period of time, Couvreur’s team synthesized squalene-based adenosine conjugates [61,62] (Figure 4D).
A completely different strategy was recently introduced by Ceglie’s team using micellar catalysis. Practically, the 1,2-epoxydodecane was coupled to 5’-phosphoribonucleotide in an aqueous cationic microemulsion. The cationic interface favored the coupling between the water insoluble epoxide and the anionic nucleotides [63].
2) Adenosine nucleolipids self-assembly
The attraction for nucleolipids in general (and adenosine nucleolipids in particular) has increased with the discovery of their capacity to form macro- or nanostructures. This unique property arises from both members of the couple: lipids have self-association ability and are known to organize in membrane layers and other structures such as liposomes, while nucleoside have base-pairing properties via hydrogen bonding or π-stacking.
Ahlers et al. were the first to explore this intriguing aspect. They compared single-chain or double-chain adenosine lipid monolayers and found that, to form stable structures, a balance had to be found between the lipophilicity procured by the alkyl chain and the size of the headgroup. Indeed, chain adenosine lipid did not form stable monolayers, while
single-Figure 4 Adenosine nucleolipids. R = lipids, R’ = H or phosphocholine. Lipids can be conjugated to
adenosine by a phosphate diester link on the primary hydroxyl group of ribose to form 5’-phosphatidyl-adenosines (A), attached to 2’,3’-hydroxyl groups of ribose through a ketal linkage (B) or ether bond (C), or coupled via an amide bond on the nucleobase (D).
A
B
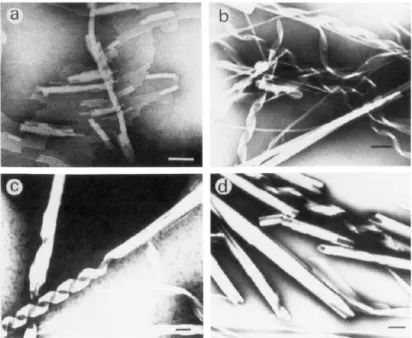
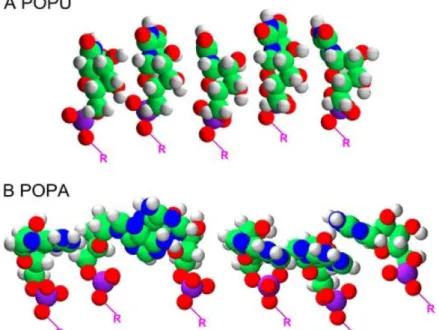
chain adenine and double-chain adenosine did [58]. Some of these adenosine nucleolipids were even found to form various helical structures, with an impact of the alkyl chain length on the efficacy of the formation of these supramolecular assemblies [55]. It was also reported that the pH of the solution greatly affected the structure: as an example, 1,2-dimyristoyl-sn-glycero-3-phosphatidyl-adenosine (DMPA) formed multihelical strands in alkaline conditions, while it formed tubular cigar-like scrolls in acidic conditions (Figure ).
Since these early explorations, adenosine nucleolipids have been described to form many different structures including micelles and liposomes (Table 2), depending on the conjugated lipid and the buffer conditions.
Figure 5 Electron micrograph of the formation process of cigar-like scrolls from DMPA in acidic solution
(pH 5.0): (a) linear double-helical strand; (b) ribbon structure; (c) incomplete cigar-like scrolls from ribbon structure; (d) complete cigar-like scrolls. All scale bars represent 0.1 µm. Reprinted with permission from [55]. Copyright 1992 American Chemical Society.
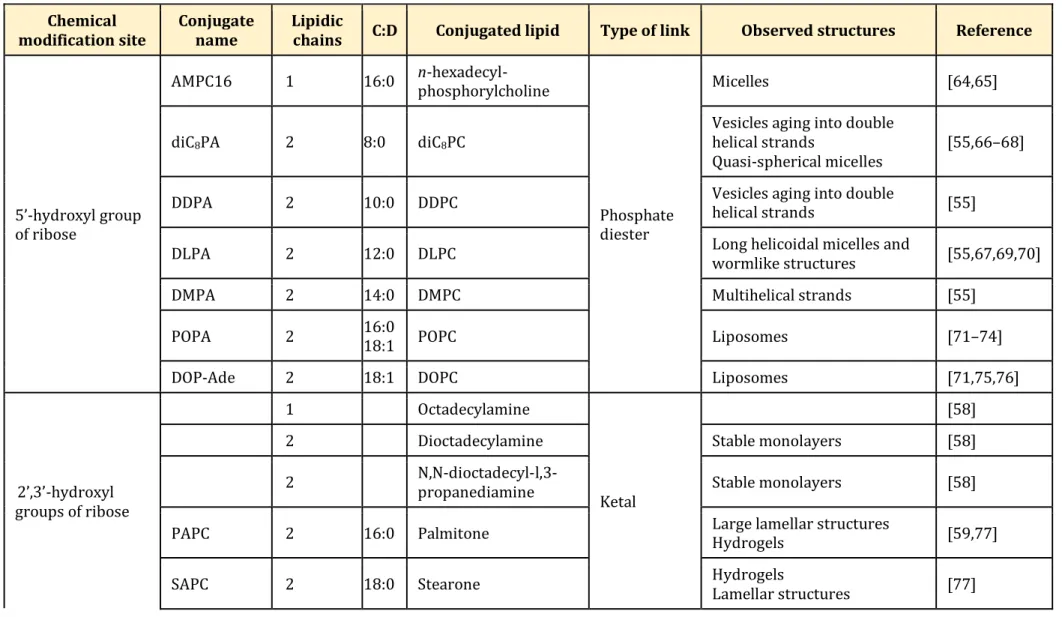
Table 2 Adenosine nucleolipids and observed macrostructures. Chemical modification site Conjugate name Lipidic
chains C:D Conjugated lipid Type of link Observed structures Reference
5’-hydroxyl group of ribose
AMPC16 1 16:0 n-hexadecyl-phosphorylcholine
Phosphate diester
Micelles [64,65]
diC8PA 2 8:0 diC8PC
Vesicles aging into double helical strands
Quasi-spherical micelles [55,66–68]
DDPA 2 10:0 DDPC Vesicles aging into double helical strands [55]
DLPA 2 12:0 DLPC Long helicoidal micelles and wormlike structures [55,67,69,70]
DMPA 2 14:0 DMPC Multihelical strands [55]
POPA 2 16:0 18:1 POPC Liposomes [71–74]
DOP-Ade 2 18:1 DOPC Liposomes [71,75,76]
2’,3’-hydroxyl groups of ribose
1 Octadecylamine
Ketal
[58]
2 Dioctadecylamine Stable monolayers [58]
2 N,N-dioctadecyl-l,3-propanediamine Stable monolayers [58]
PAPC 2 16:0 Palmitone Large lamellar structures Hydrogels [59,77]
DOAPC 2 18:1 Oleyl fatty acids Ether
Small unilamellar vesicles, worm-like structures and
giant vesicles [60] 6-amino group of adenosine SQAd 1 25:5 1,1’,2-trisnorsqualenic acid Amide Nanoparticles [61] 1 30:6 squalenylacetic-acid [78]
“C:D” represents the total amount of Carbon atoms versus the number of Double unsaturated bonds in the conjugated lipid chain. AMPC16:
adenosine 5'-hexadecylphosphate; DDPA: phosphatidyl-adenosine; DDPC: 1,2-didecanoyl-sn-glycero-3-phosphocholine; diC8PA: 1,2-dioctanoyl-sn-glycero-3-phosphatidyl-adenosine; diC8PC: 1,2-dioctanoyl-sn-glycero-3-phosphocholine; DLPA:
l,2-dilauroyl-sn-glycero-3-phosphatidyl-adenosine; DLPC: l,2-dilauroyl-sn-glycero-3-phosphocholine; DMPA:
dimyristoyl-sn-glycero-3-phosphatidyl-adenosine; DMPC: l,2-dimyristoyl-sn-glycero-3-phosphocholine; DOAPC: di-oleyl-adenosine-phosphocholine; DOP-Ade: 1,2-dioleoyl-sn-glycero-3-phosphatidyl-adenosine; DOPC: 1,2-dioleoyl-sn-glycero-3-phosphocholine; PAPC: 2′,3′-O-16-hentriacontanyliden-adenosine-5′-phosphocholine; POPA: 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidyl-adenosine; POPC: 1-palmitoyl-2-oleoyl-sn -glycero-3-phosphocholine; SAPC: 2’,3’-O-18-pentatriacontanyliden-adenosine-5’--glycero-3-phosphocholine; SQAd: squalene-adenosine.
Compared to other nucleolipids, adenosine lipids possess some special properties. In addition to forming hydrogen bonds, adenosine, like other purines, has a high stacking tendency (Table 3), which leads to a negative Gibbs free energy for adenosine-adenosine association in aqueous solution [73]. These specificities affect several structural parameters of adenosine lipid self-assemblies, thus contributing to the final supramolecular structure.
Table 3 Computed RNA nucleoside pairs stacking free energies [79]
Indeed, when comparing l,2-dilauroyl-sn-glycero-3-phosphatidyl-adenosine (DLPA) and l,2-dilauroyl-sn-glycero-3-phosphatidyl-uridine (DLPU) wormlike aggregates, Bombelli et al. reported very different behaviors upon aging: while DLPU structures remained identical, DLPA aggregates self-assembled into giant twisted helicoidal aggregates [69]. The authors attributed this dissimilarity to higher association stacking constants of adenosine compared to uridine. This property also induced tighter nucleolipid arrangement in micelles in the case of 1,2-dioctanoyl-sn-glycero-3-phosphatidyl-adenosine (diC8PA) compared to
1,2-dioctanoyl-sn-glycero-3-phosphatidyl-uridine (diC8PU) [67]. Yet, this is not the case with all adenosine
nucleolipids, as 2′,3′-O-16-hentriacontanyliden-adenosine-5′-phosphocholine (PAPC), for example, displayed a larger surface area per lipid compared to its equivalent 2′,3′-O-16-hentriacontanyliden-uridine-5′-phosphocholine (PUPC) [80].
Other phenomena attributed to adenosine high base-stacking potential were observed on several occasions by Baglioni’s team with 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidyl-adenosine (POPA) structures. First, anhydrous POPA bilayers displayed a powder-like nature, which could be explained by a specific orientation of the stacked adenosine moieties, compared to 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidyl-uridine (POPU) [73] (Figure 6). Secondly, POPA/POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) bilayers exhibited the presence of POPA-enriched microdomains, due to partial demixing of POPA and POPC [81]. The adenosine stacking interactions responsible for this behavior could be detected by hypochromism [72]. Finally, POPA/POPC vesicles were found to be more stable than the equivalent POPG (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidyl-glycerol)/POPC, where the
RNA nucleoside pair AA GG CC UU ΔGstack (kcal/mol) -2.41 -2.77 -1.76 -1.51
adenosine polar head was replaced by a glycerol head group bearing the same charge. According to the authors, only the base stacking interactions could explain this behavior [74].
Mixing adenosine nucleolipids with uridine nucleolipids can also lead to original supramolecular architectures. Indeed, besides π-stacking, the presence of a nucleoside headgroup on a nucleolipid can also drive molecular recognition through hydrogen bonding with the complementary nucleoside on another nucleolipid. A very striking example of this phenomenon was presented by Moreau et al. by mixing PAPC and PUPC. While individual nucleolipids formed large lamellar structures, mixed nucleolipids self-assembled into small vesicles which could be visualized in real-time at the micrometer scale [59]. A similar phenomenon could be observed at the macroscopic scale between 2’,3’-O-18-pentatriacontanyliden-adenosine-5’-phosphocholine (SAPC) and 2’,3’-O-18-pentatriacontanyliden-uridine-5’-phosphocholine (SUPC): when separate, these two nucleolipids form hydrogels and fluid lamellar phases above a certain temperature. When mixed, however, they form a new stable combined supramolecular system, which forms a precipitate upon heating [77].
Figure 6 Geometrical minimization for a group of POPU (A) and POPA (B) molecules performed with
Hyperchem 5.1 using AMBER force field. The orientation of purinic rings is strongly altered by the interaction between bases, whereas the pyrimidinic bases keep, more or less, their original conformation. Reprinted with permission from [73]. Copyright 2006 Elsevier.
3) Adenosine nucleolipids as delivery systems
a. Adenosine nucleolipids lipoplexes for gene deliveryAs presented above, adenosine headgroups on nucleolipids can lead to molecular recognition with complimentary nucleolipids bearing uridine headgroups. Interestingly, this phenomenon of molecular recognition was also described between adenosine headgroups on nucleolipids and nucleic acids such as RNA or DNA. As a result, self-assembled adenosine nucleolipids may be considered as promising platforms for gene delivery [82]. A great advantage of this system is that it relies on specific Watson-Crick pairings (hydrogen bonding and π-stacking) between nucleolipids and polynucleotides, offering an interesting alternative to other non-viral technologies for gene deliveries which are based on aspecific electrostatic interactions [83].
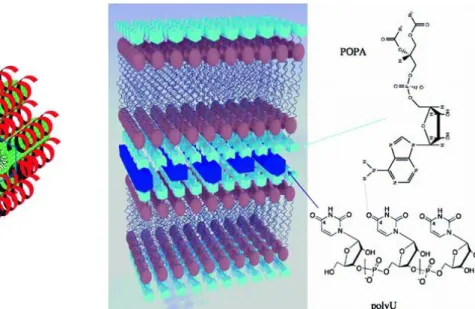
It was discovered by Baglioni’s team that when adenosine nucleolipid and polyuridilic acid (polyU) were brought together, the formation of nucleolipoplexes occurred without the presence of divalent cations (Figure 7). The proof-of-concept was obtained with diC8PA
globular micelles decorated with single-stranded complimentary RNA: this association resulted in a polynucleotide-micelle adduct that could be characterized by SAXS as a hexagonal phase (Figure 7A) [84]. Other supramolecular structures were obtained with adenosine derivatives bearing a longer lipidic chain. Indeed, POPA bilayers swollen with a polyU-containing buffer exhibited an increase in their smectic period due to the appearance of a 1D lattice of polynucleotides between the POPA lamellar stacks (Figure 7B) [85]. The formation of these nucleolipoplexes was specific to the presence of adenosine as a functional headgroup leading to selective molecular recognition of nucleic acids. For example, POPG [72,74] and POPU were shown to be less able than POPA to associate with polynucleotides [86].
Despite these interesting observations, the in vitro and in vivo proofs of concept for using these nucleolipoplexes as gene delivery systems remain extremely limited. In particular, enhanced transfection efficacies at high lipid concentrations were obtained for two uridine nucleolipids compared to conventional lipid agents (DOTAP, TransFastTM and Lipofectamine®
2000) [87,88], but so far, no study has been published regarding adenosine nucleolipids for gene delivery on pre-clinical models and no data are available in the literature regarding their transfection efficacy.
The most advanced study regarding the capacity of adenosine nucleolipid to deliver DNA inside cells was performed by Montis et al.. These authors investigated the adhesion and fusion processes of DNA-loaded POPA liposomes (formulated with POPC or 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine – DOPE – helper lipids) with giant unilamellar vesicles (GUVs – composed of POPC or 1:1=POPC:POPG) used as a model of biological cell membranes. Both nucleolipoplexes and GUV membranes could be labeled with fluorescent probes and the interaction followed by laser scanning confocal microscopy and fluorescence correlation spectroscopy. It was observed that DNA-loaded POPA liposomes entered in contact with GUVs and fused, simultaneously delivering their DNA payload. Authors claimed that they were able to reproduce in vitro a recently highlighted possible non-endocytic internalization pathway happening in vivo for nucleic acid delivery, based on the fusion of lipoplex with the cell plasma membrane [89].
For gene delivery purpose, a different approach was imagined by Arteta et al.: instead of making use of the nucleolipid self-assembling properties, these authors non-covalently decorated poly(amidoamine) (PAMAM) dendrimers with l,2-dilauroyl-sn-glycero-3-phosphatidyl-adenosine (DLPA) which has the ability to associate with nucleic acids. It is
Figure 7 Nucleolipoplexes formed by interactions between adenosine nucleolipid derivatives and
polyU. (A) Hypothetical arrangement of a hexagonal phase formed by polyU (red helixes) and cylindrical diC8PA micelles (green cylinders). Adapted with permission from [84]. Copyright 2007 John Wiley & Sons. (B) Hypothetical polyU arrangements between POPA membranes. Adapted with permission from [85]. Copyright 2007 American Chemical Society.