Relativistic Coupled Cluster theory for excited states at a general excitation rank. Applications to diatomic molecules.
204
0
0
Texte intégral
Figure

+7
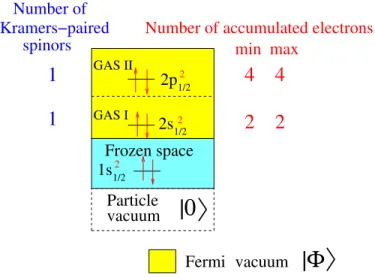
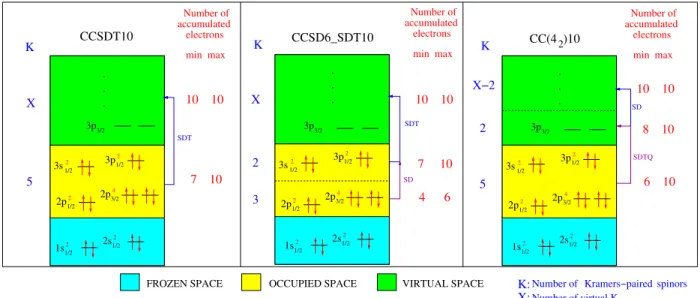

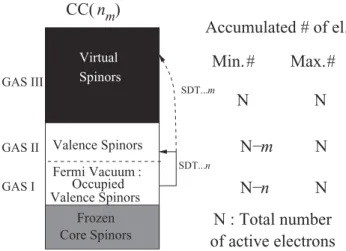
![TABLE II. Vertical excitation energies (T v ) in cm −1 for the = 1 state of the AsH molecule at the experimental bond length of R (0 e + ) ≈ R (1) e ≈ 1.5349 ˚ A [37] with different relativistic CC models (defined in Tables VIII and IX), the Dirac-Coulomb](https://thumb-eu.123doks.com/thumbv2/123doknet/14612308.732589/85.918.470.833.333.736/vertical-excitation-energies-molecule-experimental-different-relativistic-defined.webp)
Documents relatifs