The Clever Use of Pressure in Column Liquid Chromatography
7
0
0
Texte intégral
Figure
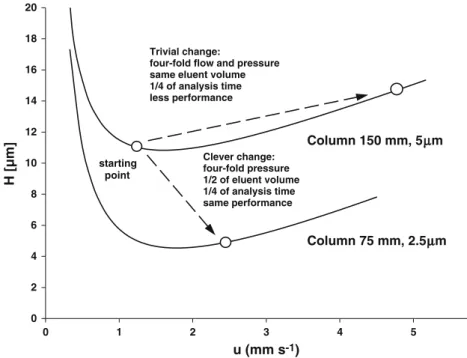
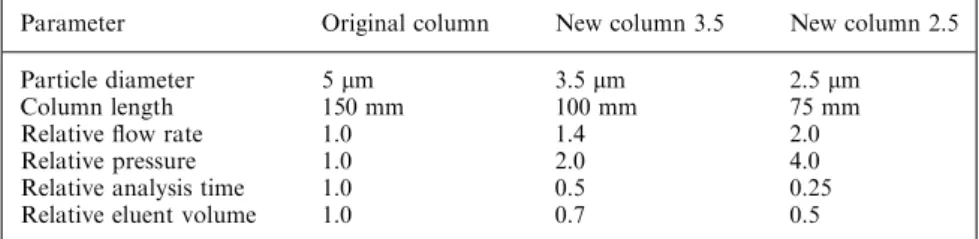
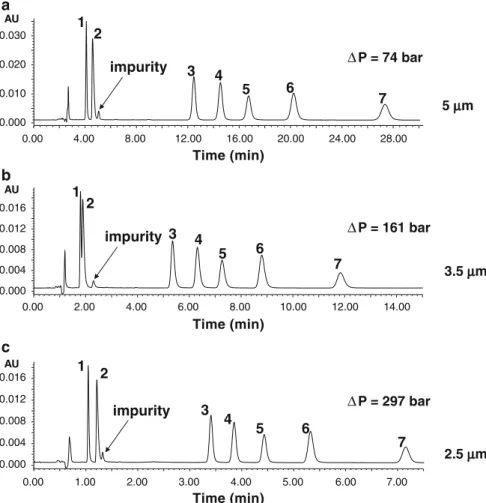
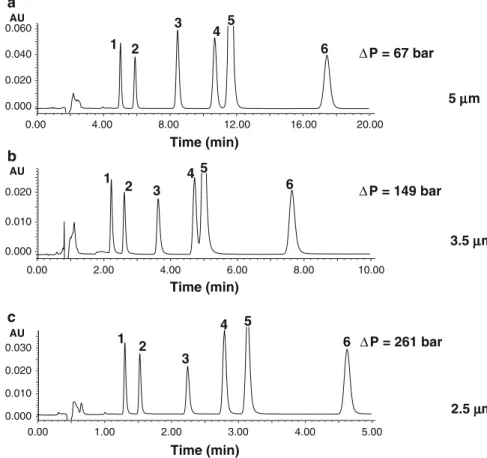
Documents relatifs