Design and Screening of Degenerate-Codon-Based
Protein Ensembles with M13 Bacteriophage
by
Griffin James Clausen
B.S. Biomedical Engineering University of Minnesota – Twin Cities, 2012
Submitted to the Department of Biological Engineering in Partial Fulfillment of the Requirements for the Degree of
Doctor of Philosophy in Biological Engineering at the
Massachusetts Institute of Technology February 2019
© 2019 Massachusetts Institute of Technology. All rights reserved.
Signature of Author: ____________________________________________________________ Griffin Clausen Department of Biological Engineering January 14, 2019
Certified by: ___________________________________________________________________ Angela Belcher James Mason Crafts Professor of Biological Engineering and Materials Science Thesis Supervisor
Accepted by: ___________________________________________________________________ Forest White Professor of Biological Engineering Graduate Program Committee Chair
This doctoral thesis has been examined by the following committee: Amy Keating
Thesis Committee Chair
Professor of Biology and Biological Engineering Massachusetts Institute of Technology
Angela Belcher Thesis Supervisor
James Mason Crafts Professor of Biological Engineering and Materials Science Massachusetts Institute of Technology
Paul Blainey
Core Member, Broad Institute
Associate Professor of Biological Engineering Massachusetts Institute of Technology
3
Design and Screening of Degenerate-Codon-Based
Protein Ensembles with M13 Bacteriophage
by
Griffin James Clausen
Submitted to the Department of Biological Engineering on January 14, 2019 in Partial Fulfillment of the
Requirements for the Degree of Doctor of Philosophy in Biological Engineering
Abstract
A billion years of evolution has crafted a diverse set of proteins capable of complex and varied functionalities. Within recent decades, scientists have applied both rational design and directed evolution to accelerate development of high-value proteins, including biotherapeutics. While computational modelling increasingly facilitates protein design, empirically screening large collections of protein variants remains an essential component of protein engineering. This process requires generating protein variation, partitioning variants with a selection pressure, and identifying highly functional proteins. This thesis presents computational tools for initial protein library design, leverages high-throughput sequencing for phage display screenings, and reports biotemplating of an inorganic phase-change material onto the filamentous M13 phage surface.
Designing ensembles of protein variants involves optimizing library size and quality with constraints on screening capacity, cost, and experimental complexity. Incorporating degenerate codons during oligonucleotide synthesis enables residue-specific protein randomization with a known amino acid scope. However, this widely adopted method often generates uneven variant abundances that diverge exponentially with additional randomized residues. The first section of this work presents tools for the design and assessment of degenerate-codon-based protein libraries. This framework facilitates incorporating an arbitrarily large number of randomized sites, non-standard genetic codes, and non-equimolar nucleotide mixtures. In addition to library size and coverage calculations, whole-population diversity metrics and abundance quantiles are reported. An evolutionary solver to optimize non-equimolar base compositions to match amino acid profiles, as well as mutational profiling for spike-in oligonucleotides is also presented.
The second section of this thesis develops an experimental and data analysis pipeline for integrating high-throughput DNA sequencing with M13 phage display biopanning. Deeply sequencing naïve M13 peptide libraries elucidated censorship patterns for both p3 and p8 coat protein fusions. Streptavidin panning recapitulated the HPQ binding motif after a single panning round, and additional biopannings pursued M13 p8 variants that interact with both gold films and carbon nanotubes. Furthermore, this thesis explores the effect of M13 p8 surface charge on the biotemplating of an inorganic phase-change material. An ambient temperature synthesis for modulating the atomic composition of germanium-tin-oxide nanomaterials is reported.
Thesis Supervisor: Prof. Angela Belcher
Acknowledgements
First and foremost, I would like to thank my advisor, Angela Belcher, for her guidance throughout my graduate experience. Angie brings a contagious enthusiasm and creativity to science that can inspire schoolchildren, graduate students, and grey-haired alumni alike. I was drawn to the breadth of research and the ambitious applications pursued by the Belcher Lab, and have learned a tremendous amount over the last few years in this unique environment.
Furthermore, I would like to thank my thesis committee members, Amy Keating and Paul Blainey for providing valuable feedback on my graduate research and guidance for my future career. I am very grateful to the members of the Belcher lab for the friendship and assistance generously shared with me. Nimrod, thank you for our countless hours of phage-centric discussion and your second-opinions on perplexing plates. Thank you, Gaelen and Rana, for helping me settle into the lab and launching the deep sequencing project. Thank you to the Bio Subgroup for the illuminating discussions and thank you, Uyanga, for organizing these valuable sessions. Thank you, Eric, for your leadership and willingness to tackle tasks that aim to benefit the broader lab. Thank you, Ngozi, for being a talented community-builder, researcher, reviewer, and ice-cream-cake-shopper. Peter, thank you for the endless conversations. Thank you, Jifa, for your assistance during my 20.109 TA-ship. Thank you, John Casey, for showing me the EHS Rep ropes and thank you, Archana, for taking on the EHS Rep ropes. Thank you, Jackie, Matt, Yu, Nurxat, Xiangnan, Neel, Alan, Will, Swati, George, Briana, Shalmalee, and everyone else!
Through my research, I had the pleasure of working with two incredibly hard-working collaborators. Thank you, Professor Desmond Loke, for leading the germanium tin oxide nanowire project as well as our Vermont hiking expedition. Thank you, Dr. Anthony Rojas from the Pentelute Lab, for the expertise and reagents you developed for our exploration of cysteine bioconjugation on phage. I was also fortunate enough to have the assistance of several bright UROPS: Jelle van der Hilst, Devany West, and Pearl Lee. Each of whom made valuable contributions towards the mutagenesis and cloning of phage, peptide conjugation, biopanning experiments, and binding validation assays.
While EHS Representative for the lab, I had the chance to engage with many diligent MIT Environment, Health, & Safety employees. Thank you for your guidance Jennifer Lynn, Hans Richter, Wilfred Mbah, Carolyn Stahl, and Roberta Polak.
5
Design and Screening of Degenerate-Codon-Based
Protein Ensembles with M13 Bacteriophage
Chapters
1 Introduction to M13 Bacteriophage, Protein Libraries, & Ligand Screening
2 Abundance Disparity and Sampling Coverage in Large Degenerate-Oligonucleotide-Based Protein Library Designs
3 High-Throughput Sequencing of Phage-Displayed Libraries and Biopanning Against Biological and Inorganic Targets
4 Biological-Templating of a Segregating Binary Alloy for Nanowire-Like Phase-Change Materials and Memory
5 Summary
Appendices
A1 General Protocols
A2 Supplementary Information for Chapter 2 A3 Supplementary Information for Chapter 3 A4 Supplementary Information for Chapter 4
Table of Contents
1 INTRODUCTION TO M13 BACTERIOPHAGE, PROTEIN LIBRARIES, & LIGAND SCREENING
Overview ... 16
M13 Bacteriophage ... 17
1.2.1 M13 Genome ... 18
1.2.2 M13 Capsid Structure ... 19
1.2.3 M13 Life Cycle with E. coli Host ... 20
Phage Display and Protein Libraries ... 22
1.3.1 Protein Randomization Techniques... 22
1.3.2 Display Technologies ... 25
1.3.3 Biopanning and Ligand Screening ... 26
Research Motivations ... 27
1.4.1 Degenerate Codon Library Design and Assessment ... 27
1.4.2 High-Throughput DNA Sequencing for Phage Display ... 29
1.4.3 M13 Biotemplating of Nanowire Materials ... 29
Structure of Thesis ... 30
References ... 32
2 ABUNDANCE DISPARITY AND SAMPLING COVERAGE IN LARGE DEGENERATE-OLIGONUCLEOTIDE-BASED PROTEIN LIBRARY DESIGNS Abstract ... 36
Introduction ... 37
2.2.1 Predicate Software and Analysis ... 38
2.2.2 Equimolar and Non-Equimolar Degenerate Codons ... 38
Results and Discussion ... 39
2.3.1 Abundance Profiles of Degenerate Oligonucleotide Ensembles ... 40
2.3.2 Abundance Quantiles for Reported NNK Peptides ... 44
2.3.3 Sampling of Protein Ensembles ... 46
2.3.4 Non-Equimolar Oligonucleotide Optimization ... 49
7
Results and Discussion – Phage Libraries... 60
3.3.1 Amino Acid and Codon Usage and Censorship ... 61
Results and Discussion – Biopanning ... 65
3.4.1 Differential Biopanning Against Streptavidin and Plastic ... 65
3.4.2 M13 p8 Library Biopanning for CNT and Gold Binders ... 69
Conclusions ... 72
References ... 73
4 BIOLOGICAL-TEMPLATING OF A SEGREGATING BINARY ALLOY FOR NANOWIRE-LIKE PHASE-CHANGE MATERIALS AND MEMORY Abstract ... 76
Introduction ... 77
4.2.1 Motivation for Ambient Synthesis of Germanium Tin Oxide Nanowires ... 77
4.2.2 Filamentous M13 Bacteriophage Surface Charge and Nanowire Biotemplating ... 79
Results and Discussion ... 80
4.3.1 Nanowire Preparation, Form, and Composition ... 80
4.3.2 WT and E3 Phage Binding Affinity to GeSnO Precursors and Bulk Oxides ... 81
4.3.3 Nanowire State-Switching and Retention Experiments ... 82
4.3.4 Laser-Pulse Excitation for Rapid Set and Reset Operations ... 84
4.3.5 Discussion of Active PCM Component in Phage-Synthesized Nanowires ... 84
Conclusion ... 85
Figures ... 87
References ... 91
5 SUMMARY Conclusions and Future Directions ... 94
References ... 97
6 APPENDIX I – GENERAL PROTOCOLS AND MATERIALS Common Stock Solutions, Media, Bacteria, and Plates ... 98
General Protocols ... 100
6.2.1 Preparation of an E. coli Overnight Culture ... 100
6.2.2 Small-Scale M13 Bacteriophage Amplification ... 100
6.2.3 Medium-Scale M13 Bacteriophage Amplification ... 101
6.2.4 Large-Scale M13 Bacteriophage Amplification ... 102
6.2.5 M13 Bacteriophage DNA Concentration Quantification by Spectroscopy ... 104
6.2.6 M13 Bacteriophage Concentration Quantification by Titering ... 104
6.2.7 Heat Shock Transformation of M13 DNA into E. coli ... 106
Phage Library Biopanning ... 107
6.3.2 Preparation of Phage DNA from Libraries and Biopannings ... 108
References ... 110
7 APPENDIX II – SUPPLEMENTARY INFORMATION FOR CHAPTER 2 IUPAC Nomenclature for Ambiguous Nucleotide Bases ... 111
Protein and DNA Library Sizes for NNK-Based Degenerate Codon Libraries ... 112
The dogma Python Package ... 113
7.3.1 Degenerate Codon Selection ... 113
Measuring Ensemble Size and Diversity ... 114
7.4.1 Entropy ... 114
7.4.2 Gini Index ... 114
7.4.3 Makowski-Soares Diversity ... 115
7.4.4 Calculated Diversity Metrics for NNK 7-mer Library using dogma ... 116
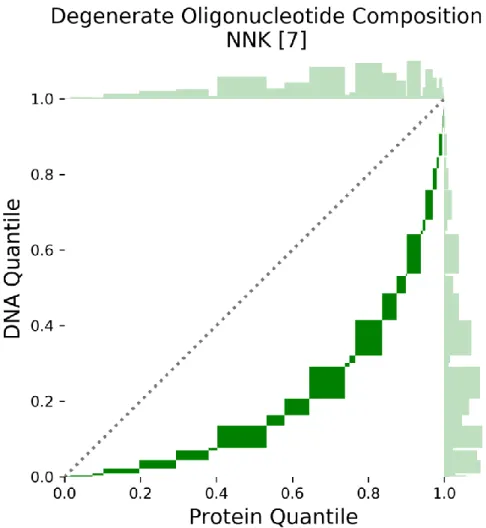
Detailed Explanation of Protein Library Quantile-Quantile Plots ... 117
The Biopanning Data Bank ... 122
Common Peptide Tags ... 123
Sampling Statistics and Library Coverage ... 124
Optimized Non-Equimolar Degenerate Codon Abundance Profile ... 125
Cost Analysis for Degenerate Oligonucleotide Synthesis Designs ... 126
Spiked Oligonucleotides for Low-Frequency, Targeted Mutations ... 127
References ... 129
8 APPENDIX III – SUPPLEMENTARY INFORMATION FOR CHAPTER 3 Protocols ... 130
8.1.1 Streptavidin Biopanning with M13 p3 Peptide Library ... 130
8.1.2 Gold Biopanning with M13 p8 Peptide Library ... 131
8.1.3 Carbon Nanotube Biopanning with M13 p8 Peptide Library ... 132
8.1.4 Preparation of Sequencing Libraries from Phage ... 133
HiSeq DNA Sample Submission ... 135
9
Acknowledgements... 145
9 APPENDIX IV – SUPPLEMENTARY INFORMATION FOR CHAPTER 4 Materials and Methods ... 146
9.1.1 Preparation of Bacteriophage ... 146
9.1.2 Synthesis of Test Suspension with Phage ... 146
9.1.3 Preparation of Test Device ... 147
9.1.4 Electrical Characterization of Device ... 147
9.1.5 Material Characterization of Test Sample ... 147
9.1.6 Binding Assay of Phage with Metal Sample ... 148
9.1.7 Optical Characterization of Suspension with Phage ... 148
9.1.8 Material Structure ... 148
Acknowledgements... 150
Supplementary Figures ... 150
List of Figures
1. Introduction to M13 Bacteriophage, Protein Libraries, & Ligand Screening
Figure 1-0 Outline for Chapter 1 ... 16
Figure 1-1 The M13KE Genome ... 18
Figure 1-2 M13 Bacteriophage Capsid Diagram... 19
Figure 1-3 M13 Bacteriophage Life Cycle with E. coli Host ... 21
Figure 1-4 Degenerate Codon Code and Amino Acid Profiles ... 24
Figure 1-5 Phage Display of Peptide Libraries Schematic ... 26
2. Abundance Disparity and Sampling Coverage in Large Degenerate-Oligonucleotide-Based Protein Library Designs Figure 2-0 Outline for Chapter 2 ... 37
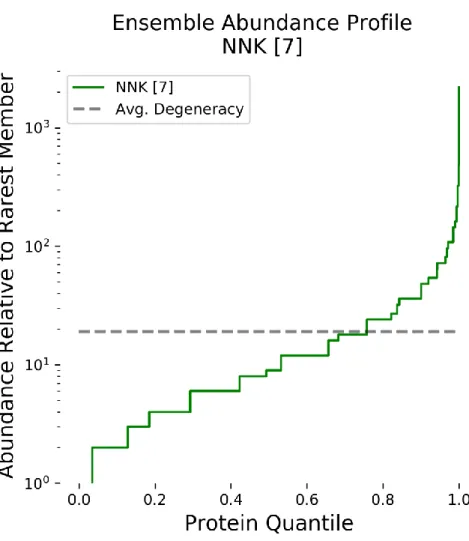
Figure 2-1 Protein Member Abundance Profile for 7-site NNK-Based Protein Library ... 41
Figure 2-2 Quantile-Quantile Plot of Degenerate Oligonucleotide with 7 NNK Codons ... 42
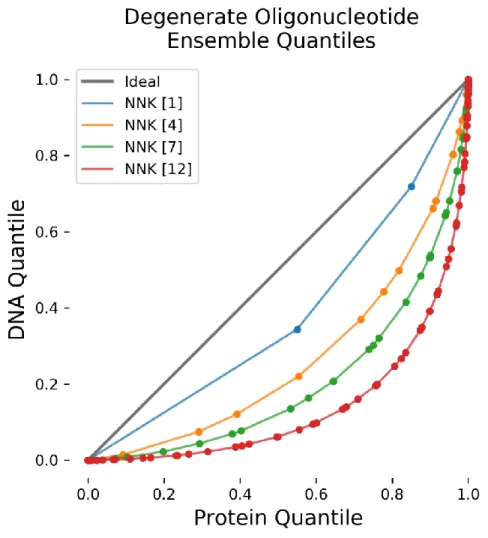
Figure 2-3 Overlay of Quantile-Quantile Plots of Various Numbers of NNK Sites ... 43
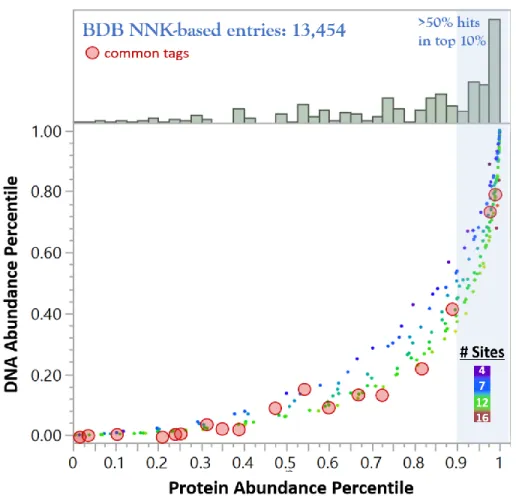
Figure 2-4 Overlay of Common Peptide Tags and Biopanning Data Bank Hits on NNK Quantile-Quantile Plot ... 45
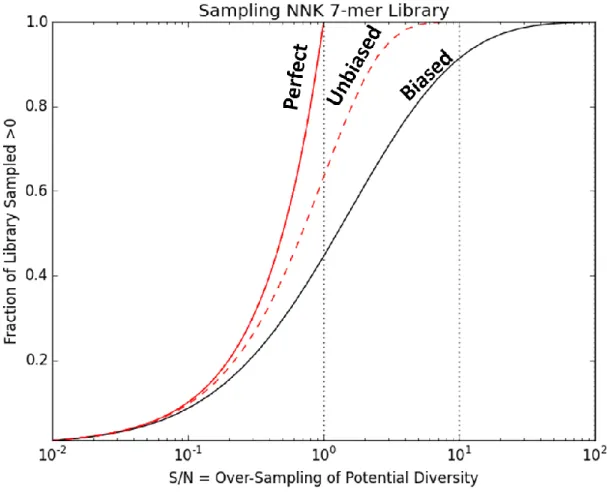
Figure 2-5 Sampling Coverage of NNK 7-mer Library for Perfect, Unbiased, and Biased Selection Models ... 48
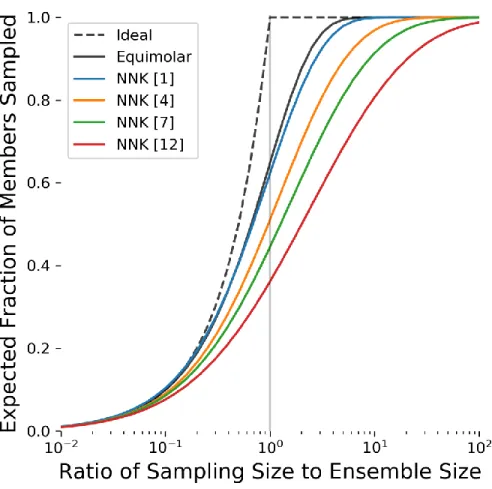
Figure 2-6 Sampling Coverage of Multiple NNK-Based Degenerate Oligonucleotide Ensembles ... 49
Figure 2-7 Overlay of Quantile-Quantile Plots of NNN, NNK, and Optimized Non-Equimolar Codon 7-mers ... 51
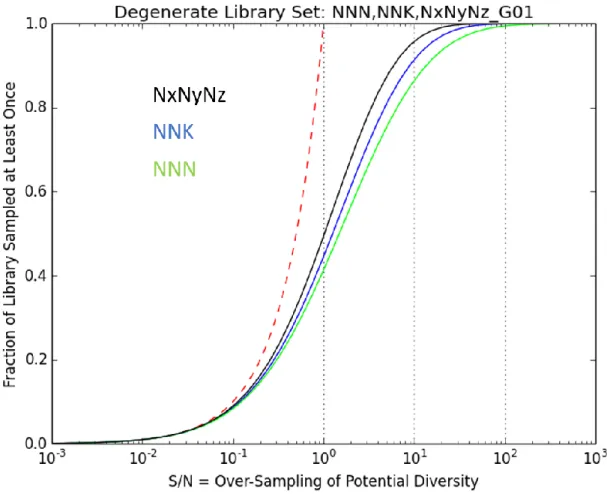
Figure 2-8 Overlay of NNN, NNK, and NxNyNz 7-site Library Sampling Coverages ... 52
Appendix II Figure A2-1 Degenerate Codon Efficiency Landscape... 113
Figure A2-2 Graphical Example of Degenerate Codon Selection with dogma ... 114
Figure A2-3 Gini Index as a Measure of Ensemble Diversity ... 115
Figure A2-4 Diversity Metrics for NNK Libraries of Various Lengths ... 116
Figure A2-5 Block Quantile-Quantile Plot of NNK Library ... 117
Figure A2-6 Block Quantile-Quantile Plot for NNK-2 Library ... 118
11
3. High-Throughput Sequencing of Phage-Displayed Libraries and Biopanning Against Biological and Inorganic Targets
Figure 3-0 Outline for Chapter 3 ... 57
Figure 3-1 Distribution of p8 8-mer Peptides by Number of Positive and Negative Residues ... 64
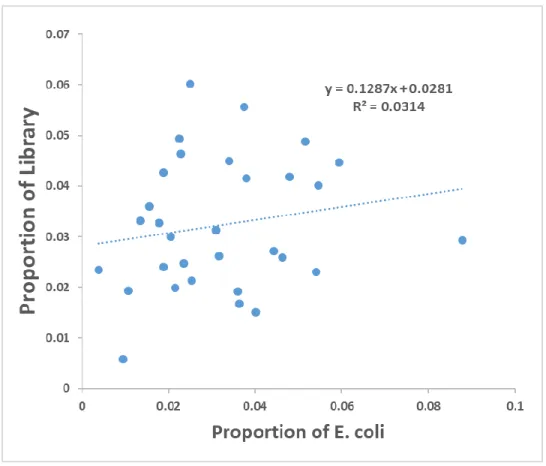
Figure 3-2 Comparison of Codon Use Proportions in M13 p3 Library and E. coli ... 65
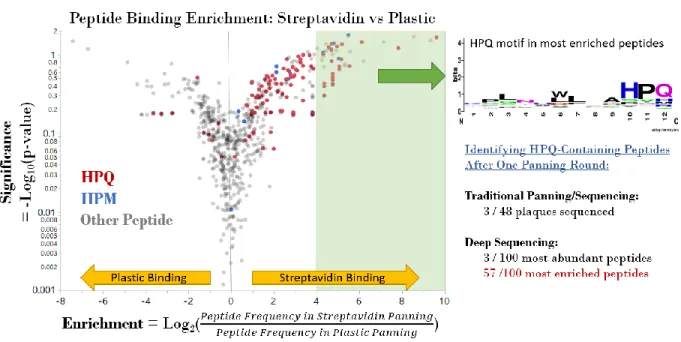
Figure 3-3 Peptide Binding Enrichment Results for Streptavidin Panning ... 67
Figure 3-4 NNK Quantiles of Top 1,000 Peptides in Naïve Libraries and Streptavidin Panning ... 68
Figure 3-5 Peptide Binding Enrichment Results for Carbon Nanotube Panning ... 70
Figure 3-6 Peptide Binding Enrichment Results for Gold Film Panning ... 71
Appendix III Figure A3-1 Theoretical Distribution of Peptides by Number of Positive and Negative Residues in 8-mer GNK-NNK7 Library ... 141
Figure A3-2 Comparison of Codon Use Ranks in M13 p3 Library and E. coli ... 142
Figure A3-3 PSSM for Linear 12-mer Peptide Pannings Against Streptavidin; Rounds 1 and 2 ... 143
Figure A3-4 Distributions of Insert Lengths from p3 12-mer Streptavidin Biopanning ... 144
Figure A3-5 ELISA Binding Assay for Streptavidin Biopanning Hits ... 145
4. Biological-Templating of a Segregating Binary Alloy for Nanowire-Like Phase-Change Materials and Memory Figure 4-0 Outline for Chapter 4 ... 77
Figure 4-1 Binding, Synthesis and Utility of the Nanoscale, Wire-Like Phase-Change-Based Material with Phage . 87 Figure 4-2 Control and Tuning Phase-Change-Based Material Components via Phage-Enabled Binding ... 88
Figure 4-3 Investigation of the Mechanism and Characteristics of the Phage-Enabled Binding for the Phase-Change-Based Material ... 89
Figure 4-4 Phage-Templated PCM Structures Exhibit Controllable and Reliable Switching Characteristics ... 90
Appendix IV Figure A4-1 Transmission Electron Microscopy (TEM) Images of the Sample with WT Phage... 150
Figure A4-2 Energy Dispersive X-ray Spectroscopy (EDS) Mapping of the Sample with WT Phage ... 151
Figure A4-3 UV-Vis Absorption Spectroscopy Spectra of Suspensions with Varying Synthesis Temperatures ... 151
Figure A4-4 Dependence of the Suspension Absorbance on the Heating Time ... 152
Figure A4-5 UV-Vis Absorption Spectroscopy Spectra of the Suspensions from Varying Synthesis Duration ... 153
Figure A4-6 Dependence of the E3 Sample Resistance on the Laser Exposure Time ... 154
Figure A4-7 X-Ray Diffraction (XRD) Patterns of the Sample with No Phage (top) and E3 Phage (bottom) ... 155
Figure A4-8 XRD Patterns of the Sample with E3 Phage Before (top) and After (bottom) Laser Exposure ... 156
Figure A4-9 Pulsed Current-Voltage Characteristics of Sample with E3 Phage ... 157
Figure A4-11 Memory Characteristics of the Sample Without Phage ... 158 Figure A4-12 Dependence of E3 Sample Resistance on Applied Pulse Length Under Electric Field Conditions ... 159 Figure A4-13 Schematic of the Electrical Measurement Setup ... 159
13
List of Tables
Table 1-1 M13 Bacteriophage Genes and Gene Products ... 20
Table 3-1 Sequencing Overview for Naïve Phage-Displayed Peptide Libraries ... 60
Table 3-2 M13 p3 Linear-7 Library Amino Acid Usage ... 62
Table 3-3 M13 p8 Linear-8 Library Amino Acid Usage – Hyun-Jung’s Library ... 63
Table A1-1 Table of Dilutions and Expected Plaques for Titering ... 104
Table A2-1 IUPAC Nomenclature for Ambiguous Nucleotide Bases ... 111
Table A2-2 Protein and DNA Library Sizes for NNK-Based Degenerate Codon Libraries ... 112
Table A2-3 Degeneracy Table for 7-mer NNK Library under supE Translation ... 121
Table A2-4 Common Peptide Tags ... 123
Table A2-5 Cost Analysis for Degenerate Oligonucleotide Synthesis ... 126
Table A3-1 DNA Preparation and Barcode Matching for HiSeq Submissions ... 135
Table A3-2 M13 p3 C7C Library Amino Acid Usage ... 136
Table A3-3 M13 p3 Linear-7 Library Amino Acid Usage ... 137
Table A3-4 M13 p8 Linear-8 Library Amino Acid Usage – Paul’s Library ... 138
Table A3-5 M13 p8 Linear-8 Library Amino Acid Usage – Hyun-Jung’s Library ... 139
Table A3-6 M13 p3 Linear-12 Library Amino Acid Usage ... 140
List of Acronyms and Abbreviations
MeaningA269 Absorbance at 269 nm
BDB Biopanning Data Bank bp Base pairs
BSA Bovine serum albumin CFU Colony-forming unit CVD Chemical-vapor deposition
Da Daltons
DC Degenerate codon
DREME Discriminative Regular Expression Motif Elicitation dsDNA Double-stranded DNA
eDC Equimolar degenerate codon (equal ratios of nucleotides present in mixture) ELISA Enzyme-linked immunosorbent assay
IUPAC International Union of Pure and Applied Chemistry MOI Multiplicity of infection
15 PBS Phosphate-buffered saline PCM Phase-change material
PCR Polymerase chain reaction PFU Plaque-forming unit PhD Phage display
PP Polypropylene
PS Packaging signal; Polystyrene QQ-Plot Quantile-quantile plot
RF Replicative form RT Room temperature SBA Segregating binary alloy scFv Single-chain variable fragment ssDNA Single-stranded DNA
TBS Tris-buffered saline
1
Introduction to M13 Bacteriophage, Protein Libraries, & Ligand
Screening
Overview
M13 bacteriophage is an elegantly simple virus and a broadly useful biotechnology tool that serves as a unifying thread for this body of work. To lay the common foundation for subsequent chapters, this section first presents fundamental information about the M13 genome, capsid structure, and life cycle. Next, the concepts of protein libraries and ligand screening are introduced within the context M13 phage display. Finally, a brief overview of the research projects that comprise this thesis is presented and shared themes and motivations discussed. This work includes an analysis of the common protein library generation strategy for phage display; methodology for high-throughput DNA sequencing of phage libraries and biopanning against both inorganic nanomaterials and proteins; and the electrostatically driven formation of an inorganic oxide onto the filamentous capsid of M13 p8 variants.
17
M13 Bacteriophage
Bacteriophages, or phages, are a diverse class of viruses that infect bacteria to generate progeny. Present from the depths of the oceans to the heights of the atmosphere, phages are estimated to outnumber bacteria 10:1 globally.1, 2 With an estimated worldwide population of approximately 1031 particles, bacteriophages are the most abundant biological entities in the
biosphere.3 Yet, such ubiquity of phages is not exclusive to our natural environment. Since Frederick William Twort first identified bacteriophages in 1915, scientists have employed phages in the laboratory to generate fundamental insights into molecular biology.4 Landmark phage-enabled discoveries include confirmation of DNA as the genetic material by Hershey and Chase,5 the quantification of nucleotides in a codon,6 and the discovery of restriction enzymes.7
Isolated from sewers in Munich, Germany in 1963 by Hofschenider,8 M13 bacteriophages have become widely used in molecular biology and biotechnology laboratories. During the late 1970s, M13 was demonstrated to be a versatile DNA cloning vector.9,10 And in the mid-1980s, George Smith introduced the technique of ‘phage display’ using M13, demonstrating that phage with a foreign DNA fragment inserted into a capsid-encoding gene could be isolated using an antibody reactive to the displayed foreign protein sequence.11 Applications of M13 phage display are diverse and include epitope-mapping and ligand discovery.12 As evidence of the impact made by M13 phage display, George Smith and Sir Gregory Winter were awarded half of the 2018 Nobel Prize in Chemistry for this pioneering work.
The concept of M13 phage display has been further expanded to study interactions between proteins and inorganic materials,13,14 and taking advantage of the filamentous nanostructure of M13 as a template to create functional nanomaterials.15,16 Understanding the M13 genome, capsid structure, and infection lifecycle is important for engineering viable M13 variants and fusion libraries, and for assessing censorship at both the amino acid and codon levels.
1.2.1 M13 Genome
The M13 bacteriophage genome is approximately 7 kilobases in size and in a single-stranded, circular form when packed into the M13 capsid [Figure 1-1]. Nine genes are contained in the M13 genome, encoding eleven proteins that enable intracellular DNA replication, the formation of a membrane secretion pore, and comprise the virion capsid [Table 1-1]. In addition, the genome contains an origin of replication and a packaging signal required for protein capsid assembly around the ssDNA. Overall, M13 exhibits greater than 97% DNA sequence homology between the other Ff class filamentous phage, f1 and fd.17
Several widely used M13 vectors have been optimized for DNA cloning and phage display experiments. The M13mp18 vector and M13KE vector are engineered variants of M13 that incorporate a LacZ selectable marker and a multiple cloning site (MCS) to facilitate DNA cloning. In particular, to facilitate the N-terminal peptide fusions on the major coat protein p8, M13KE has been refactored to separate the overlapping coding and regulatory elements of p8 and p9.18
19 1.2.2 M13 Capsid Structure
In contrast to icosahedral and prolate geometries of many notable bacteriophages, such as T4 and λ phage, the capsid of M13 is an elongated helical form.19 The single-stranded genome is packed within the capsid core, and the length of the virion is dependent on genome size.20 Laboratory strains of M13 are filamentous structures, approximately 900 nm in length and 6.5 nm in diameter.19 This high aspect ratio of ~140:1 is akin to a 4-foot long pencil [Figure 1-2].
The body of the M13 capsid is composed of approximately 2,700 copies of the p8 major coat protein. This 50 amino acid helical protein is arranged in an overlapping helically pitched cylinder around the ssDNA genome.21 The core region of each p8 forms hydrophobic interactions with neighboring capsid proteins and only several N-terminal amino acids are solvent-exposed. Both ends of the M13 filament are capped with 3-5 pairs of minor coat proteins. Five copies of p7 and p9 interact with the DNA packaging signal to form the ‘head’ or ‘distal tip’ of the virion. Five copies of p3 and p6 proteins similarly cap the ‘tail’ of M13. P3 is the largest capsid protein and consists of three distinct domains. The N-terminal domain, N1, initiates cytosolic translocation of the ssDNA genome during infection. The middle domain, N2, enables binding to the F pilus on the E. coli surface. The C-terminal domain, CT, interfaces with the p6 and p8 proteins.22
Figure 1-2 M13 Bacteriophage Capsid Diagram
This diagram depicts the composition of the M13 bacteriophage capsid. The “tail” of M13 consists of 3-5 p6 and multi-domain p3 proteins. The bulk of the virus is composed of p8 proteins that helically wrap around the encapsulated ssDNA M13 genome. The “head” of M13 is composed of several p7 and p9 capsid proteins. The true aspect ratio of M13 (~140:1) is approximately 20x longer than depicted in this diagram.
Table 1-1 M13 Bacteriophage Genes and Gene Products
The nine genes of M13 bacteriophage encode for eleven proteins, including two truncated gene products. Six proteins are required for DNA replication and bacteriophage assembly, and five proteins form the mature bacteriophage capsid. Amino acid counts and MW from Russel et al. (2004).22 Note: Roman and Arabic numerals are used interchangeably for M13 genes and gene products.
Gene Protein Primary Function Amino Acids MW [Da] #/Virion g1 p1 Membrane pore, regulator 348 39,502 -
g2 p2 DNA Endonuclease 410 46,137 -
g3 p3 Minor capsid protein, host recognition 406 42,522 5 g4 p4 Membrane pore component 405 43,476 -
g5 p5 ssDNA sequestration 87 9,682 -
g6 p6 Minor capsid protein 112 12,342 5
g7 p7 Minor capsid protein 33 3,599 5
g8 p8 Major capsid protein 50 5,235 ~2,700
g9 p9 Minor capsid protein 32 3,650 5
g10 p10 DNA replication, regulator 111 12,672 - g11 p11 Membrane pore component 108 12,424 -
1.2.3 M13 Life Cycle with E. coli Host
M13 is a Group II (single-stranded DNA) virus of the family Inoviridae, genus Inovirus, and species Enterobacteria phage M13, and member of the Ff class of filamentous viruses.23 The host bacteria for M13 and other Ff phage are Escherichia coli harboring the F conjugative episome. M13 infection of E. coli is non-lytic and non-temperate, resulting in a chronic state of infection. Infection slows bacterial growth rate and viral protein synthesis accounts for approximately 30% of host metabolic activity.22
21
replicative form (RF). As additional RF DNA copies are replicated, the encoded viral proteins are transcribed and translated by host machinery, generating cytoplasmic and membrane-bound viral proteins.
Once sufficient p5 has been translated, p5 dimers sequester newly synthesized M13 ssDNA. Complexes of p7 and p9 proteins bind to the secondary structure of the M13 DNA packaging signal and initiate extrusion through membrane pore complexes formed by p1, p4 and p11. Major coat proteins embedded within the host’s inner membrane displace p5 proteins on the ssDNA as the head of the phage exits the cell. After p8 capsid monomers have coated the entire genome, p3 and p6 proteins cap the tail of the phage and the virion is released into the extracellular environment. Approximately 1,000 progeny phage are released during primary cellular infection, although the host can continue to extrude phage at lower rate or become re-infected.22
Figure 1-3 M13 Bacteriophage Life Cycle with E. coli Host
M13 infection is initiated upon p3 binding to the tip of the host F pilus. As the pili retract, p3 domains interact with Tol proteins in the host membrane. M13 capsid proteins are integrated into the membrane as the ssDNA is transferred into the cytoplasm. Host enzymes convert the ssDNA to a double-stranded replicative form (RF) and M13 genes are expressed. Progeny ssDNAs are synthesized by rolling cycle amplification and sequestered by p5 dimers. M13 gene products for a membrane pore complex extending both membranes and capsid proteins are translated into the inner host membrane. Minor coat proteins p7 and p9 interact with the ssDNA packaging signal (PS) and the pore complex. As the ssDNA is translocated from the cytoplasm to the extracellular space, the p5 dimers are removed and p8 proteins surround the DNA. Finally, p6 and p3 proteins cap the virion structure as the DNA is fully extruded, releasing a mature M13 progeny.
Phage Display and Protein Libraries
During the 1970s, M13-based vectors were developed with restriction enzyme cut-sites and selectable markers amenable to amplifying foreign DNA fragments ligated into non-protein-encoding regions of the genome.10 M13 vectors can be replicated in E. coli, such that ssDNA can be isolated from purified phage or dsDNA harvested from infected bacteria. [Figure 1-3] It wasn’t until 1985 that M13 was genetically engineered to express foreign protein fragments onto native capsid proteins. This ‘phage display’ technique was first demonstrated by George Smith, who inserted a foreign DNA into a capsid-encoding gene, and isolated the chimeric virions using an antibody reactive to the encoded foreign protein sequence.11
The incorporation of mixed (or degenerate) oligonucleotides cloned into phage capsid proteins led to the parallel synthesis of phage with different foreign DNA insertions and expressing different fusion proteins, thus generating libraries of protein variants amenable to screening experiments. Notable applications of phage display screening include epitope mapping,12 protease profiling,24 and early-stage drug discovery.25 Notably, phage display was employed to discover the antibody fragment D2E7, which was used to develop the blockbuster anti-inflammatory drug adalimumab (Humira®).26 M13 phage display is also frequently used to isolate novel high-affinity peptide ligands from libraries of random peptides.27
1.3.1 Protein Randomization Techniques
Protein libraries, such as those screened using phage display, can be generated from a variety of random mutagenesis, focused mutagenesis, and recombination strategies.28 Random mutagenesis methods, such as error-prone PCR (epPCR) or the use of mutator strains, facilitate the accumulation of randomly distributed mutations throughout a DNA region.29 Notably,
23
Focused mutagenesis allows researchers to restrict the scope and position of amino acid variation, thus facilitating a more targeted exploration of the vast theoretical protein space. For low-throughput and highly focused experiments, DNA sequences for each protein variant can be predefined and synthesized individually in tubes or plates. Microarrays containing 100,000s of features are feasible but are costly and oligo length is limited to 60 nt, too short for many M13 phage display applications.31 Alternatively, modern DNA synthesizers can readily synthesize DNA libraries many orders of magnitude larger by simultaneously adding multiple bases during step-wise oligo synthesis.32 Such DNA libraries are comprised of individual DNA variants with abundances determined by the composition of bases added during synthesis.
Cloning DNA libraries created by degenerate oligonucleotide synthesis is the predominant method for generating protein libraries for phage display.22 Most commonly, equal proportions of mixed bases are used, with each of the (42-1) = 15 possible equimolar combinations of the four
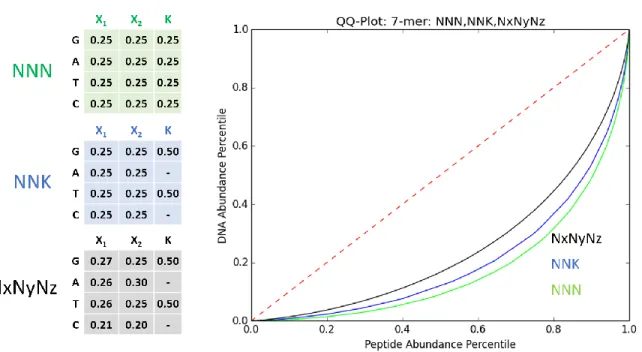
standard DNA bases A, C, G, and T assigned a single-letter code by IUPAC [Figure 1-4]. A triplet of degenerate bases is called a degenerate codon (DC). For example, the codon NNN (where N = A, C, G, or T) comprises all 64 possible standard codons, and the codon NNK (where K = G or T) encompasses 32 possible codons. Both NNN and NNK designs encompass codons that encode all 20 standard amino acids, thus they are referred to as ‘hard-randomizing’ DCs. Additional degenerate codon schemes that encode useful sets of amino acids have been compiled.33 Furthermore, non-equimolar mixtures of bases are readily synthesized within 1% precision. This facilitates degenerate codon optimization to approximate specified amino acid profiles,34 or control low-frequency mutations in a large protein region, akin to a targeted epPCR.35
The protein library composition resulting from a degenerate codon library depends on the genetic code used for translation. For phage display, it is common to use amber (UAG) stop codon suppressor E. coli strains (supE/glnV). SupE strains can translate UAG stop codons into glutamine, thus preventing any stop codons for NNK randomized sites. Regardless of the translation system, most DCs encode for amino acids at uneven frequencies. [Figure 1-4]. One alternative to DCs is trinucleotide cassettes during oligo synthesis, enabling a single codon to be specified for each desired amino acid; however, this method is over ten times as costly as DCs and requires long manufacturing lead time.36 Another approach to mitigate amino acid bias is to
combine multiple DCs. The 22c-Trick37 and related methods require multiple mutagenic primers to be combined at specified ratios to balance amino acid composition; however, such methods are not conducive to randomizing multiple contiguous or nearby residues, which is desired in peptide library preparation.
Figure 1-4 Degenerate Codon Code and Amino Acid Profiles
The notation for ambiguous DNA bases was standardized by IUPAC in 1970 (left). Shaded entries indicate the nucleotide bases (column) present in the specified IUPAC letter code (row). See Table A2-01 for verbose table with complementary codes. Nucleotide triplets containing at least one degenerate base are called degenerate codons. The degeneracy, or redundancy, of an amino acid refers to the number of unique codons by which it is encoded. The supE translation system suppresses the amber stop codon (UAG) with glutamine (Q). The amino acid abundance of two common hard-randomization degenerate codons NNK and NNN are presented where N = (A or C or G or T) and K = (G or T).
DCs enable a cost-effective and flexible method for generating arbitrarily large protein libraries. As multiple residues are simultaneously randomized, the theoretical diversity of the resulting protein library expands exponentially, rapidly exceeding the ability to physically create.
25 1.3.2 Display Technologies
M13 phage display was the original platform for display-based screening, but additional technologies have also been developed; these include yeast display,38 bacteria display,39 mRNA display,40 and ribosome display.41 Furthermore, non-M13 phages including T442 and T743 have also been pursued for display-based experiments. Each display platform has certain advantages and disadvantages, but each shares the core capacity to physically link a testable phenotype (displayed protein fusion) with an easily determined genotype (encoding DNA). Determination of which display platform to use depends on the protein scaffold to be randomized, the diversity of the envisioned library, and laboratory capabilities.
Both mRNA and ribosome display are unencumbered by DNA transformation into cells, thus library sizes approaching 1014 protein variants can be synthesized. However, the highly
technical and time-consuming protocols have limited wide-spread adoption outside of pioneering labs.44 Yeast display enables eukaryotic expression as well as quantitative screening with fluorescent-activated cell sorting. Although cellular transformation limits yeast display to libraries of ~108, yeast display has become particularly valuable for antibody engineering and for
proteins requiring complex folding or post-translational modifications.
Phage display is particularly suited for the high-throughput screening of short peptides and small protein domains such as Kunitz domains and scFvs.45, 46 Furthermore, commercial phage display kits facilitate widespread adoption with minimal laboratory barriers or specialized training. Although all five M13 capsid proteins have been used as fusion proteins, the most commonly used capsid proteins are p3 and p8.22 The viability of p8 peptide fusions dramatically decreases with size, with 8 amino acid peptide fusions commonly used as the upper limit.47 The viability, or reduced infectivity, of p8 particular amino acid fusion sequences varies and is not easily predicted a priori.48 In contrast, the low valency p3 capsid protein can accommodate large fusions of greater than 100 amino acids [Figure 1-5].49 This dissertation focuses on M13 phage libraries expressed on the high valency p8 major coat protein and on the low valency p3 minor coat protein.
Figure 1-5 Phage Display of Peptide Libraries Schematic
This figure depicts the parallel construction of phage variants expressing random peptides on the N-terminal of p3 minor coat proteins. The base M13 genome is first digested by restriction enzymes flanking the desired insert region. Short DNA oligonucleotides are either synthesized or digested to have complementary overhangs and are ligated into the cut phage genome. DNA oligos can encode a specific protein sequence or can consist of an ensemble of random oligonucleotides, often generated using degenerate codon synthesis. After transformation into E. coli, the resulting phage with displayed peptides are generated and can be purified and used for subsequent screening experiments.
1.3.3 Biopanning and Ligand Screening
Biopanning is the process of iteratively screening a library of phage-displayed proteins to enrich for a variant (or group of variants) that has a high affinity for an immobilized target.
27
conducted without replicates or negative controls, and the most abundant binding peptides are identified using low-throughput Sanger sequencing. Modern DNA sequencing technology can provide much deeper insight into the composition of large and diverse peptide libraries before and after screening, providing millions versus dozens of DNA reads to the assess the relative abundance of peptides.
Research Motivations
The purpose of this work is to enhance and apply the methods by which M13 bacteriophage is used to discover novel peptide ligands to biological and inorganic materials. This begins with the development of a Python package dogma that calculates the complete abundance profile of degenerate-oligonucleotide-based protein libraries, such as the common 7-, 8-, and 12-site NNK libraries used in phage display and other protein engineering experiments. Furthermore, high-throughput DNA sequencing has revolutionized the characterization of phage display experiments. Deep sequencing of naïve M13 peptide libraries and differential panning methodologies are needed to assess M13 censorship, particularly of p8 libraries, and to avoid experimental sources of error that enrich for non-target-related peptides. Finally, the ability to control the amino acid composition on the surface of the filamentous M13 virion structure provides the possibility of modulating the atomic composition of biotemplated inorganic materials. To demonstrate this capability, and to synthesize a nanowire material that is incompatible with traditional high-temperature processing, M13 variants with different p8 major coat proteins were used to guide the synthesis of a germanium-tin-oxide nanomaterial.
1.4.1 Degenerate Codon Library Design and Assessment
Incorporation of degenerate codons (DCs) is the most widely used strategy for achieving site-specific control of amino acid randomization, particularly for phage display libraries.50 Most DCs, especially hard-randomizing codons (NNN, NNK, NNS, NNB), encode amino acids unevenly due to the redundant nature of the genetic code mapping codons to amino acids. It is widely known that this imbalance increases exponentially as additional residues are randomized. For an NNK 12-mer library, each of the most abundant proteins is 531,441 times more frequent than
each of the rarest members. Not only might such abundance disparity handicap discovery of rare and valuable variants during a screening experiment, but perhaps more importantly, the rare variants are more likely to be excluded from a screen altogether during the library creation or subsampling stages. Despite this, libraries with many random residues, whether contiguous or distributed within a scaffold protein structure, are frequently employed in order to explore a broader portion of the theoretical protein space. Notably, in a recent review of non-immunoglobulin protein scaffolds being developed for biotherapeutic discovery, over half of the 20 reported protein library scaffolds, such as Fynomers and Alphabodies, included between 6-17 NNK randomization sites.51
Accurately accounting for this initial abundance imbalance is important for comparing alternative randomization designs, determining the desired extent of randomization, estimating the number of unique variants sampled for a screening experiment, and assessing the ability of a screening method to identify rare but functional variants. Previously reported computational programs that characterize protein library size and the expected library sampling coverage are limited in several ways. Constraints include a restrictive DC limit of six codons, use confined to the standard genetic code, and inability to accommodate non-equimolar proportions of nucleotide bases.52 The most commonly used phage display protocols use suppressor strains of E. coli and involve more than six DCs. Further, commercial DNA synthesis vendors such as IDT® provide non-equimolar mixed bases, allowing for further optimization of protein library composition. Thus, more practical tools to assess degenerate codon library designs are needed. In particular, computer programs to further facilitate degenerate codon selection for non-standard genetic codes, an arbitrary number of random sites, and non-equimolar bases are required.
29
mutational profiling of spike-in oligonucleotides. Specifically, a non-equimolar codon composition was designed to produce a more equitable amino acid profile; thus, generating a more balanced protein library and increasing unique members represented for the same sampling effort and cost.
1.4.2 High-Throughput DNA Sequencing for Phage Display
Standard phage display biopanning consists of iteratively screening upwards of 1012
viruses, then ascertaining the DNA sequence identities of dozens, perhaps hundreds of enriched hits. This process of individually amplifying phage, extracting DNA, and Sanger sequencing is a bottleneck and requires many rounds of panning and subsequent amplification. High-throughput sequencing changes this paradigm by facilitating millions of reads at a fraction of the cost. However, experimental methods are needed to process collections of phages into short DNA fragments amenable to NGS sequencing and to analyze the resulting data.
The key research goals of Chapter 3 are to establish methods for deep sequencing both p3 and p8 peptide libraries, recapitulate a known streptavidin peptide motif using NGS without repetitious panning rounds, and subsequently conduct p8 biopanning against inorganic nanomaterials used as in vivo imaging probes. Upon sequencing naïve libraries, the extent of amino acid censorship in p3 and p8 phage display peptide libraries were investigated, as were codon-level bias and position-specific expression patterns. Furthermore, these NNK-based peptide libraries were assessed to validate whether the peptide abundances largely followed the theoretical abundance profile.
1.4.3 M13 Biotemplating of Nanowire Materials
Extending beyond its role as a DNA cloning vector and a passive vehicle for protein library screening, M13 phage has also demonstrated value in its capacity to bind and nucleate growth of inorganic materials on the nanoscale. Motivated by the ability of natural proteins to control nucleation and organization of inorganic materials such as abalone shells,53 the filamentous structure of M13 provides a modular scaffold for the synthesis of a variety of nanomaterials including high-surface area meshes for catalysis and zinc-sulfide nanofibers for battery anodes.54
Germanium-tin-oxide, as a phase-change material (PCM), is a promising candidate for achieving universal memory in computers.55 PCMs retain information based on reversible transitions between physical states with contrasting physical properties. Germanium-tin-oxide is capable of switching between amorphous and crystalline states that have drastically different electrical conductivities. Because germanium-tin-oxide is impossible to synthesize in the necessary nanowire form using standard CVD methods (the atomic components segregate at high temperatures), alternative synthesis approaches are worth exploring.56
The key research objective for pursued in Chapter 4 is to demonstrate the feasibility of ambient temperature biotemplating of germanium and tin onto the surface of M13 to generate nanomaterials capable of switching between high- and low-resistive states. Furthermore, M13 p8 variants with additional glutamic acids expressed on the major coat proteins were compared to demonstrate that both atomic composition and subsequent device performance could be modulated. This work explores the control of morphology, composition and functionality of SBA-based PCMs using template-directed nucleation achieved via electrostatic-binding selectivity of the M13 bacteriophage surface.
Structure of Thesis
This introductory chapter has presented a brief overview of the foundational knowledge and motivations for the research topics explored in subsequent sections. The remainder of this thesis addresses these aforementioned research projects in three main chapters.
Chapter 2, Abundance Disparity and Sampling Coverage in Large Degenerate-Oligonucleotide-Based Protein Library Designs, presents the software package dogma and its
31
and processing sequencing data are presented. Multiple naïve peptide libraries displayed on p3 and p8 are sequenced to ascertain broad censorship biases at the codon and amino acid level, to identify overly abundant peptide clones, and to generate a database of viable M13 variants for future reference. Subsequently, differential biopanning methods were developed for screening peptide libraries against streptavidin as a proof-of-principle and then applied to biopanning for both carbon nanotubes and gold nanoparticles. Protocols and supplemental information for this chapter is found in Appendix III.
Chapter 4, Biological-Templating of a Segregating Binary Alloy for Nanowire-Like Phase-Change Materials and Memory, is largely reproduced, with permission, from a manuscript published in ACS Applied Nano Materials.57 The synthesis of germanium-tin-oxide nanowires onto the surface of M13 is presented, followed by an analysis of material morphology, composition, and device performance. Main text figures are grouped in a separate subsection at the end of the chapter and supplementary information is recorded in Appendix IV.
Finally, Chapter 5, Summary, ties together the major results presented in the subsequent chapters with future extensions. Additional information is contained in Appendices. Appendix I contains general protocols and information regarding standard solutions and lab materials. Appendix II, III, and IV contain chapter-specific supplementary information for the three main research projects.
References
[1] Bergh, Ø., BØrsheim, K. Y., Bratbak, G., and Heldal, M. (1989) High abundance of viruses found in aquatic environments, Nature 340, 467.
[2] Reche, I., D’Orta, G., Mladenov, N., Winget, D. M., and Suttle, C. A. (2018) Deposition rates of viruses and bacteria above the atmospheric boundary layer, The ISME Journal 12, 1154. [3] Clokie, M. R., Millard, A. D., Letarov, A. V., and Heaphy, S. (2011) Phages in nature,
Bacteriophage 1, 31-45.
[4] Cairns, J., and Delbrück, M. (1966) Phage and the origins of molecular biology, Cold Spring Harbor Laboratory of Quantitative Biology.
[5] Hershey, A. D., and Chase, M. (1952) Independent functions of viral protein and nucleic acid in growth of bacteriophage, The Journal of General Physiology 36, 39-56.
[6] Crick, F., Barnett, L., Brenner, S., and Watts-Tobin, R. J. (1961) General nature of the genetic code for proteins, Nature.
[7] Luria, S. E., and Human, M. L. (1952) A nonhereditary, host-induced variation of bacterial viruses, Journal of Bacteriology 64, 557.
[8] Hofschneider, P. v., and Preuss, A. (1963) M13 Bacteriophage Liberation from Intact Bacteria as Revealed by Electron Microscopy, Journal of Molecular Biology 7, 450-451.
[9] Ray, D. S., and Kook, K. (1978) Insertion of the Tn3 transposon into the genome of the single-stranded DNA phage M13, Gene 4, 109-119.
[10] Griffin, H. G., and Griffin, A. M. (1993) DNA sequencing, Applied Biochemistry and Biotechnology 38, 147.
[11] Smith, G. P. (1985) Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface., Science 228, 1315-1317.
[12] Dieltjens, T., Willems, B., Coppens, S., Nieuwenhove, L. V., Humbert, M., Dietrich, U., Heyndrickx, L., Vanham, G., and Janssens, W. (2010) Unravelling the antigenic landscape of the HIV-1 subtype A envelope of an individual with broad cross-neutralizing antibodies using phage display peptide libraries., Journal of Virological Methods.
[13] Whaley, S. R., English, D. S., Hu, E. L., Barbara, P. F., and Belcher, A. M. (2000) Selection of peptides with semiconductor binding specificity for directed nanocrystal assembly., Nature 405.
[14] Lee, S. W., Mao, C., Flynn, C. E., and Belcher, A. M. (2002) Ordering of quantum dots using genetically engineered viruses., Science 296.
[15] Nam, K. T., Kim, D.-W., Yoo, P. J., Chiang, C.-Y., Meethong, N., Hammond, P. T., Chiang, Y.-M., and Belcher, A. M. (2006) Virus-enabled synthesis and assembly of nanowires for
33
[18] Ghosh, D., Kohli, A. G., Moser, F., Endy, D., and Belcher, A. M. (2012) Refactored M13 bacteriophage as a platform for tumor cell imaging and drug delivery, ACS Synthetic Biology 1, 576-582.
[19] Marvin, D. a., Symmons, M. F., and Straus, S. K. (2014) Structure and assembly of filamentous bacteriophages, Progress in Biophysics and Molecular Biology, 1-43.
[20] Rakonjac, J., Feng, J.-n., and Model, P. (1999) Filamentous phage are released from the bacterial membrane by a two-step mechanism involving a short C-terminal fragment of pIII, Journal of Molecular Biology 289, 1253-1265.
[21] Overman, S. A., Tsuboi, M., and Thomas Jr, G. J. (1996) Subunit Orientation in the Filamentous Virus Ff (fd, f1, M13), Journal of Molecular Biology.
[22] Russel, M., Lowman, H. B., and Clackson, T. (2004) Introduction to phage biology and phage display, Phage Display: A Practical Approach, 332.
[23] Marvin, D., Hale, R., Nave, C., and Citterich, M. H. (1994) Molecular models and structural comparisons of native and mutant class I filamentous bacteriophages: Ff (fd, f1, M13), If1 and IKe, Journal of Molecular Biology 235, 260-286.
[24] Ratnikov, B., Cieplak, P., and Smith, J. W. (2009) High throughput substrate phage display for protease profiling, In Proteases and Cancer, pp 93-114, Springer.
[25] Nixon, A. E., Sexton, D. J., and Ladner, R. C. (2014) Drugs derived from phage display: from candidate identification to clinical practice, MAbs 6, 73-85.
[26] Salfeld, J., Kaymakcalan, Z., Tracey, D., Roberts, A., and Kamen, R. (1998) Generation of fully human anti-TNF antibody D2E7, Arthritis Rheumatism 41, S57.
[27] Kay, B. K., Adey, N. B., He, Y. S., Manfredi, J. P., Mataragnon, A. H., and Fowlkes, D. M. (1993) An M13 phage library displaying random 38-amino-acid peptides as a source of novel sequences with affinity to selected targets., Gene 128.
[28] Packer, M. S., and Liu, D. R. (2015) Methods for the directed evolution of proteins, Nature Reviews Genetics 16, 379.
[29] Zhao, H., Giver, L., Shao, Z., Affholter, J. A., and Arnold, F. H. (1998) Molecular evolution by staggered extension process (StEP) in vitro recombination, Nature Biotechnology 16, 258. [30] Vanhercke, T., Ampe, C., Tirry, L., and Denolf, P. (2005) Reducing mutational bias in random
protein libraries, Analytical Biochemistry 339, 9-14.
[31] Kosuri, S., and Church, G. M. (2014) Large-scale de novo DNA synthesis: technologies and applications., Nature Methods 11, 499-507.
[32] Hill, D. E., Oliphant, A. R., and Struhl, K. (1987) Mutagenesis with degenerate oligonucleotides: An efficient method for saturating a defined DNA region with base pair substitutions, In Methods in Enzymology, pp 558-568, Elsevier.
[33] Tonikian, R., Zhang, Y., Boone, C., and Sidhu, S. S. (2007) Identifying specificity profiles for peptide recognition modules from phage-displayed peptide libraries., Nature Protocols 2, 1368-1386.
[34] Labean, T. H., and Kauffman, S. A. (1993) Design of synthetic gene libraries encoding random sequence proteins with desired ensemble characteristics, Protein Science 2, 1249-1254. [35] Cárcamo, E., Roldán-Salgado, A., Osuna, J., Bello-Sanmartin, I. n., Yáñez, J. A., Saab-Rincón,
G., Viadiu, H. c., and Gaytán, P. (2017) Spiked Genes: A Method to Introduce Random Point Nucleotide Mutations Evenly throughout an Entire Gene Using a Complete Set of Spiked Oligonucleotides for the Assembly, ACS Omega 2, 3183-3191.
[36] Krumpe, L. R. H., Schumacher, K. M., McMahon, J. B., Makowski, L., and Mori, T. (2007) Trinucleotide cassettes increase diversity of T7 phage-displayed peptide library., BMC Biotechnology 7, 65.
[37] Kille, S., Acevedo-Rocha, C. G., Parra, L. P., Zhang, Z. G., Opperman, D. J., Reetz, M. T., and Acevedo, J. P. (2013) Reducing codon redundancy and screening effort of combinatorial protein libraries created by saturation mutagenesis, ACS Synthetic Biology 2, 83-92. [38] Boder, E. T., and Wittrup, K. D. (2000) Yeast surface display for directed evolution of protein
expression, affinity, and stability, In Methods in Enzymology, pp 430-444, Elsevier. [39] Daugherty, P. S. (2007) Protein engineering with bacterial display, Current Opinion in
Structural Biology 17, 474-480.
[40] Wilson, D. S., Keefe, A. D., and Szostak, J. W. (2001) The use of mRNA display to select high-affinity protein-binding peptides, Proceedings of the National Academy of Sciences 98, 3750-3755.
[41] Hanes, J., and Pluckthun, a. (1997) In vitro selection and evolution of functional proteins by using ribosome display., Proceedings of the National Academy of Sciences 94, 4937-4942. [42] Malys, N., Chang, D. Y., Baumann, R. G., Xie, D., and Black, L. W. (2002) A bipartite bacteriophage T4 SOC and HOC randomized peptide display library: detection and analysis of phage T4 terminase (gp17) and late sigma factor (gp55) interaction., Journal of Molecular Biology 319.
[43] Danner, S., and Belasco, J. G. (2001) T7 phage display: a novel genetic selection system for cloning RNA-binding proteins from cDNA libraries., Proceedings of the National Academy of Sciences 98.
[44] Barendt, P. A., Ng, D. T., McQuade, C. N., and Sarkar, C. A. (2013) Streamlined protocol for mRNA display, ACS Combinatorial Science 15, 77-81.
[45] Dennis, M. S., and Lazarus, R. A. (1994) Kunitz domain inhibitors of tissue factor-factor VIIa. I. Potent inhibitors selected from libraries by phage display., J Biol Chem 269.
[46] Griffiths, A. D., Williams, S. C., Hartley, O., Tomlinson, I. M., Waterhouse, P., Crosby, W. L., Kontermann, R. E., Jones, P. T., Low, N. M., and Allison, T. J. (1994) Isolation of high affinity human antibodies directly from large synthetic repertoires., Embo J 13.
[47] Iannolo, G., Minenkova, O., Gonfloni, S., Castagnoli, L., and Cesareni, G. (1997) Construction, exploitation and evolution of a new peptide library displayed at high density by fusion to the major coat protein of filamentous phage., Biol Chem 378.
[48] Kuzmicheva, G. A., Jayanna, P. K., Sorokulova, I. B., and Petrenko, V. A. (2008) Diversity and censoring of landscape phage libraries., Protein Eng Des Sel.
35
[53] Belcher, A. M., Wu, X., Christensen, R., Hansma, P., Stucky, G., and Morse, D. (1996) Control of crystal phase switching and orientation by soluble mollusc-shell proteins, Nature 381, 56.
[54] Zhang, G., Wei, S., and Belcher, A. M. (2018) Biotemplated Zinc Sulfide Nanofibers as Anode Materials for Sodium-Ion Batteries, ACS Applied Nano Materials 1, 5631-5639.
[55] Xiong, F., Liao, A. D., Estrada, D., and Pop, E. (2011) Low-power switching of phase-change materials with carbon nanotube electrodes, Science 332, 568-570.
[56] Yu, B., Ju, S., Sun, X., Ng, G., Nguyen, T. D., Meyyappan, M., and Janes, D. B. (2007) Indium selenide nanowire phase-change memory, Applied Physics Letters 91, 133119.
[57] Loke, D., Clausen, G., Ohmura, J., Chong, T.-C., and Belcher, A. (2018) Biological-Templating of a Segregating Binary Alloy for Nanowire-Like Phase-Change Materials and Memory, ACS Applied Nano Materials.
2
Abundance Disparity and Sampling Coverage in Large
Degenerate-Oligonucleotide-Based Protein Library Designs
Abstract
Incorporating mixtures of bases during step-wise oligonucleotide synthesis, often according to degenerate codon patterns, enables site-specific protein randomization with a defined scope of amino acids. This strategy provides a simple and broadly used method for generating vast libraries of protein variants. However, many degenerate codon schemes encode amino acids unevenly, and the disparity between rare and abundant protein variants increases exponentially as multiple residues are randomized. Accurately accounting for this initial library imbalance is important for comparing alternative randomization designs, accurately estimating the number of variants sampled in a screen, and assessing the ability of a screening method to identify rare but functional variants.
To address these needs, this work presents a suite of computational tools, dogma, for the design and assessment of degenerate-oligonucleotide-based protein ensembles. This software expands upon previous library design tools to accommodate libraries with an arbitrarily large
37
to minimize protein variant disparity and increasing library richness for the same sampling effort. Furthermore, dogma is used to retrospectively assess the initial library quantile, or normalized rank, of published peptide hits in the Biopanning Data Bank repository of biopanning results. Finally, dogma is used to calculate mutation rates and amino acid compositions for spiked-in libraries, wherein small proportions of nucleotides are mixed throughout a template oligo to generate distributed mutations throughout a large protein region, akin to error-prone PCR.
Figure 2-0 Outline for Chapter 2
Introduction
In recent decades, both rational design and directed evolution have successfully accelerated the development of novel high-value proteins, including biotherapeutics. While computational modeling increasingly facilitates protein design, empirically screening protein libraries remains an essential component of protein engineering.1 Synthesis of oligonucleotides containing degenerate codons is a low-cost and widely used technique for generating ensembles of random DNA that can be translated into libraries of protein variants for screening.2 However, many degenerate codons encode unequal proportions of amino acids due to the redundancy of the genetic code, and protein variant abundances can become exceedingly uneven as additional degenerate codons are incorporated.3 This imbalance increases exponentially as additional residues are randomized, such that, for an NNK 12-site library (where N = A, C, G, or T and K = G or T mixed bases), each of most abundant proteins is 531,441 times more abundant than each of the rarest members. Not only does such disparity handicap valuable, yet rare, variants during a screening experiment, more importantly, the rare variants are more like to be excluded a screen altogether during the library creation or subsampling stages.4
However, libraries with many randomized residues, whether contiguous or distributed within a scaffold protein structure, are frequently used with the hope of exploring a broader portion of the theoretical protein space. Notably, in a recent review of non-immunoglobulin protein scaffolds being developed for biotherapeutic discovery, over half of the 20 reported library scaffolds, such as Fynomers and Alphabodies, included between 6-17 NNK randomization sites.5 Such bias in naïve libraries has important implications for determining the expected number of unique proteins present in a sample drawn from a library, as well as assessing the ability of rare variants to be positively selected under a given screening paradigm.
2.2.1 Predicate Software and Analysis
Many library design tools have been published to facilitate protein library design. Software and web servers such as LibDesign,6 MDC Analyzer,7 SwiftLib,8 LibraryDesign,9 and DYNAMCC10, 11 provide algorithms for selecting one or more equimolar degenerate codons that efficiently encode a specified set of amino acids for randomized residues. In an equimolar degenerate codon, each of the nucleotides present in a particular base is evenly represented. Incorporating multiple degenerate codons with reduced amino acid scopes can focus screening efforts and minimize or even eliminate bias induced by the genetic code.12 However, these approaches are not applicable to consecutive randomized sites, such as for peptide library designs.
Several web servers have also been published specifically to calculate library coverage statistics for degenerate codon library analysis, namely GLUE-IT and TopLib.4, 13 Applicability of these web servers is restricted to systems using the standard genetic code, limited in the number of degenerate positions, exclusive to equimolar degenerate codons, and these tools do not
39
degenerate codons (eDC), each comprising a unique set of standard codons. The composition of amino acids that derive from each degenerate codon depends on the translation system employed, and tables of commonly used equimolar degenerate codons have been reported.16 Non-equimolar degenerate codons (neDC), where uneven mixtures of nucleotides are combined, have also been studied and are amenable to standard oligo synthesis. Low-percentages of mixed-bases throughout a large template oligonucleotide, akin to error-prone PCR, has been pursued to optimize enzymatic and activities.17, 18 Furthermore, several optimization algorithms based on gradient descent or grid search methods have sought to find non-equimolar base profiles to encode target amino acid compositions at single residue positions.19, 20 For standard mixed base synthesis with the 1% precision, there are 176,851 possible unique combinations of A, C, G, and T proportions. Thus, there are over 5.5e15 possible non-equimolar degenerate codon compositions. The object-oriented codebase of dogma was further extended to perform both non-equimolar codon optimization and simulate mutation profiles for spike-in libraries.
Results and Discussion
A Python package named dogma was written in an object-oriented manner to perform stable and efficient calculations for large degenerate-oligonucleotide-based protein libraries. The source code for dogma is available at http://github.com/griffinclausen/dogma or can be installed as a Python package dogma through PyPi https://pypi.org/project/dogma/. As an extension to the core dogma codebase, a simplified degenerate codon selection tool was created to accommodate custom genetic code definitions and filtering based on exclusion and inclusion of specified codons and amino acids [Figure A2-1, Figure A2-2]. To support sequence motif discovery algorithms, including DREME21, that use a list of control sequences in order to identify enriched motifs within a subset of sequences. Dogma provides a facile interface for generating an arbitrarily large in silico library of representative protein or DNA sequences from any degenerate codon library using the samples method for Oligonucleotide objects.