Dynamics of Rod-Coil Block Copolymers
MASSACHUSETTS INSTITUTE OF TECHNOLOLGY Muzhou Wang
JUN
16
2015
B.S., California Institute of Technology, 2009
LIBRARIES
Submitted to the Department of Chemical Engineering in partial fulfillment of the requirements for the degree of
Doctor of Philosophy at the
Massachusetts Institute of Technology
August, 2014
LSepe1r
2,014K C 2014 Massachusetts Institute of Technology. All rights reserved.Signature of Author:
Signature redacted
Department of Ienl Engineering August, 2014
Certified by:
Signature redacted
Bradley D. Olsen Paul M. Cook Career Development Assistant Professor of Chemical Engineering
Thpcie 4Zmrervisor
Signature redacted
Accepted by:
Patrick S. Doyle Professor of Chemical Engineering Chairman, Committee for Graduate Students
Dynamics of rod-coil block copolymers
Muzhou Wang
Submitted to the Department of Chemical Engineering on August 25, 2014 in partial fulfillment of the requirements of the degree of
Doctor of Philosophy in Chemical Engineering Abstract
Polymer science is exploring advanced materials which combine functional domains such as proteins and semiconducting polymers with traditional flexible polymers in the same molecule. While thermodynamic assemblies of different geometries introduce many interesting new phenomena such as entropic packing and liquid crystalline interactions, dynamic effects are also important to understand for optimal design of material mechanics, processing kinetics, and the new physics that directly arises from the motion of multiple domains of dissimilar geometries. Rod-coil block copolymers are an example of hybrid molecules that have attracted recent interest for organic electronics and biomaterials.
This thesis uses theory, simulation, and experiments to better understand the mechanisms of molecular motion in entangled rod-coil block copolymers as a model for this wider class of functional polymeric materials. The large geometrical differences between rigid rods and Gaussian coils cause significant nonlinearities in dynamical behavior as these two motifs are combined on the same molecule. It is shown that the motion of rod-coils is slowed relative to rod and coil homopolymers because of a mismatch between the curvature of the rod and coil entanglement tubes. This mismatch causes different relaxation mechanisms in the small and large rod limits, where either the rod or the coil block is expected to determine the motion of the overall molecule. In the small rod limit where the rod is a perturbation on coil motion, the randomly varying curvature of the coil's tube presents entropic barriers to the reptation of the rod, modifying the unhindered motion of the coil along its tube into an activated reptation process. In the large rod limit where the coil is a perturbation on rod motion, the long rod cannot rotate around the surrounding entanglements so motion is only possible when the coil moves into a straightened entanglement tube in an arm retraction process.
These mechanisms were first explored in molecular dynamics simulation using the Kremer-Grest model of entangled polymers, which showed that the tracer diffusion of coil-rod-coil triblock copolymers was slower than both rod and coil homopolymers. This observation was verified experimentally by forced Rayleigh scattering in both the small and large rod limits. Using additional simulations and experiments, the theories were applied to more complex configurations such as diblock copolymers and self-diffusion in polymer melts, where additional effects including molecular asymmetry and the motion of the surrounding chains were incorporated. Finally, a more detailed and quantitative analysis of the mechanisms both individually and in combination was performed using a coarse-grained slip-link model which matched the previous simulations and experiments.
Thesis supervisor: Bradley D. Olsen, Paul M. Cook Career Development Assistant Professor of Chemical Engineering
Acknowledgements
My accomplishments in graduate school and any future success will always be indebted to so many people.
It has been a privilege and pleasure to be one of Brad's first students. He is a very dedicated mentor, providing me with freedom, support, and hours upon days of advice. I was lucky enough to know him since my undergrad years, and I hope it has been a joy watching me grow from a nafve college junior. My time with him has undoubtedly made me a better scientist. I am particularly grateful for the experience of starting a new research program from square one, and I
look forward to hearing about his accomplishments in the future. Don't forget to take it easy. One of the things I will miss most about MIT is my time with the Olsen group. Matt Glassman has been a friend since undergrad, and is one of the most thoughtful, conscientious, and useful researchers I have ever met. Shengchang Tang is a great source of amusement, literature, and general feedback about any topic. Other members of the constantly growing group have all helped me in many ways, including Chris, Michelle, Yin, Charlotte, Vinay, Dongsook, Reggie, Aaron, Bokyung, Yan, Minkyu, and Manos, and I wish I had more time with the newest members. I am so glad to have worked with such amazing colleagues, and also great friends. Please keep in touch. The future of polymer science is in great hands.
I have also had excellent collaborators during my time here. Alfredo Alexander-Katz taught me the first things to know about molecular simulation. Alexei Likhtman is an incredible expert on entangled polymer dynamics, willing to share seemingly endless results and knowledge.
I have also worked with very talented undergrads, Gina Noh and Ksenia Timachova. I am sure they will be very successful at UC Berkeley and beyond.
Thanks also to Prof. Armstrong and Bazant, who rounded out my thesis committee and provided excellent advice over the years. Many other members of MIT ChemE and PPST were helpful, especially as our research group was starting up. My teaching experience with Karthik, Arup, and Brad were also particularly enjoyable.
We are grateful for our funding sources from the MIT Research Support Committee, DuPont, and the National Science Foundation. Fellowships from the American Society for Engineering Education and the National Science Foundation have allowed the academic freedom to pursue interesting problems. Computational resources were generously provided by Prof. Alan Hatton's group and XSEDE/Teragrid.
Special thanks to my friends and family for their support. I wouldn't be where I am without my parents, who have been great sources of support and encouragement. Others have been crucial in times of need and fun in times of enjoyment, especially Nathan and Jon.
And finally, I will be forever grateful to my soon-to-be wife Kara. My time at MIT would never have been the same without her. Life ahead will bring many challenges and adventures, but I am always at peace knowing that she will be by my side. This thesis is dedicated to her, but only if she reads it in full. Otherwise, it goes to Lazarus and Jesus, who are currently basking on a rock.
Table of Contents
A cknow ledgem ents ... 6
List of Figures... 10
List of Schem es... 13
List of Tables ... 13
Chapter 1: Background ... 14
1.1 Introduction... 14
1.2 Rod-Coil Block Copolym ers... 16
1.3 Theories of Polym er Dynam ics ... 18
1.4 M easurem ents by Experim ents and Sim ulation... 21
1.5 Thesis Overview ... ... 24
1.6 References... 25
Chapter 2: M ethods... 30
2.1 Synthesis of Rod-Coil Block Copolym ers... 30
2.1.1 Synthesis of poly(ethylene glycol)-b-polyalanine copolymers... 30
2.1.2 Synthesis of poly(alkoxyphenylene vinylene)-b-polyisoprene copolymers ... 38
2.2 Forced Rayleigh Scattering... 46
2.2.1 Theoretical background ... 47
2.2.2 Experim ental setup and data processing ... 52
2.2.3 o-nitrostilbene dye ... 60
2.2.4 D ithienylethene dye ... 66
2.3 Krem er-Grest M olecular Dynam ics Sim ulation ... 72
2.3.1 Theoretical background ... 73
2.3.2 Im plem entation ... 77
2.3.3 Equilibration ... 85
2.4 Coarse-grained Slip-link Sim ulation... 87
2.4.1 Theoretical background ... 88
2.4.2 Im plem entation ... 91
2.5 References... 100
Chapter 3: Rouse D ynam ics of Rod-Coil Block Copolym ers ... 102
3.1 Background... 102
3.2 Problem Statem ent... 103
3.3 Brownian Dynam ics Sim ulation... 104
3.4 Sm oluchow ski Form ulation... 106
3.5 Single Isotropic Bead Approxim ation ... 110
3.6 Conclusions... 115
3.7 References... 116
Chapter 4: D iffusion of Entangled Rod-Coil Block Copolym ers... 118
4.1 A bstract... 118
4.2 M ain Body ... 119
4.3 References... 129
Chapter 5: Experimental measurement of coil-rod-coil block copolymer tracer diffusion through entangled coil hom opolym ers ... 132
5.3 Experim ental... 135
5.4 Results and D iscussion ... 142
5.5 Conclusions... 153
5.6 References... 153
Chapter 6: Diffusion mechanisms of entangled rod-coil diblock copolymers ... 156
6.1 Abstract... 156
6.2 Introduction... 157
6.3 M ethods... 160
6.4 Results and D iscussion ... 163
6.5 Conclusions... 176
6.6 References... 177
Chapter 7: Self-diffusion and constraint release in isotropic entangled rod-coil block copolymers ... 180
7.1 Abstract... 180
7.2 Introduction... 181
7.3 Experim ental... 183
7.4 Results and D iscussion ... 188
7.5 Conclusions... 202
7.6 References... 203
Chapter 8: Tube curvature slows motion of rod-coil block copolymers through activated reptation ... 206
8.1 Abstract... 206
8.2 M ain Body ... 207
8.3 References... 217
Chapter 9: Crossover between activated reptation and arm retraction mechanisms in entangled rod-coil block copolym ers ... 220
9.1 Abstract... 220
9.2 Introduction... 221
9.3 Slip-Link M odel of Rod-Coil Block Copolym ers ... 224
9.4 Curvilinear D iffusion in Activated Reptation... 228
9.5 Com bining A ctivated Reptation and Arm Retraction... 232
9.6 3-D im ensional Center-of-M ass Diffusion ... 243
9.7 Conclusions... 245
9.8 References... 246
Chapter 10: Conclusions... 248
10.1 Sum m ary ... 248
10.2 Outlook ... 251
Appendix A : Supporting Inform ation for Chapter 4... 254
Appendix B: Supporting Inform ation for Chapter 5... 260
Appendix C: Supporting Inform ation for Chapter 6... 268
Appendix D : Supporting Inform ation for Chapter 7... 270
Appendix E: Supporting Inform ation for Chapter 8... 280
List of Figures
1-1. Tube m odel schem atic... 19
2-1. Synthesis of dye-labeled PEO-b-PA coil-rod-coil triblock copolymers ... 31
2-2. Construction of polypeptide genes ... 33
2-3. Sequence and electrophoresis of polypeptide genes and proteins... 35
2-4. Anion exchange chromatography of PEO-B3 reaction... 37
2-5. Synthesis of dye-labeled PEO-b-PA rod-coil diblock copolymers ... 38
2-6. 'H-NMR spectrum of 2,5-di(2'-ethylhexyloxy)toluene ... 40
2-7. TLC of 2,5-di(2'-ethylhexyloxy)-4-methylbenzaldehyde synthesis ... 42
2-8. 'H-NMR spectrum of 2,5-di(2'-ethylhexyloxy)-4-methylbenzaldehyde ... 42
2-9. 'H-NMR spectrum of poly(2,5-di(2'-ethylhexyloxy)-1,4-phenylene vinylene)...44
2-10. Thin layer chromatography of PPV-b-PI synthesis... 46
2-11. Coherent beams crossing on sample in FRS experiment ... 47
2-12. Schematic of optics for FRS experiment... 54
2-13. Brass sample cell design for FRS experiment...57
2-14. Sam ple FR S results... 59
2-15. 1H-NMR spectrum of o-nitro-p-tolunitrile ... 62
2-16. 'H-NMR spectrum of ONS-CN...63
2-17. 'H-NMR spectrum of ONS-COOH... 65
2-18. 'H-NMR spectrum of ONS-M...66
2-19. Gel permeation chromatography of PPV-b-PI synthesis and irradiation ... 68
2-20. 1H-NMR spectrum of acetyl-DTE... 70
2-21. 1H-NMR spectrum of vinyl-DTE ... 72
2-22. Probability distribution of adjacent monomer separation in Kremer-Grest model ... 74
2-23. C ell list algorithm ... 81
2-24. Aggregation of rod homopolymers due to depletion interactions ... 82
2-25. Simulation parallel computation efficiency... 84
2-26. Excluded volume potentials in slow push-off equilibration method...86
2-27. Equilibration of Kremer-Grest simulation... 87
2-28. Slip-link model schematic ... 89
2-29. Matching MD and slip-link for coil homopolymers... 92
2-30. Matching MD and slip-link for mean-squared displacement of rod homopolymers ... 94
2-31. Matching MD and slip-link for rotational correlation of rod homopolymers ... 95
2-32. Matching MD and slip-link for rod-coil block copolymers ... 96
2-33. Comparing time step size in slip-link model...98
2-34. Slip-link density and extension over time ... 100
3-1. Mean-squared displacement of dilute solution rod-coil block copolymer ... 106
3-2. Diffusion of rod-coils in dilute solution ... 106
3-3. Diffusion of rod-coils compared to Smoluchowski prediction ... 109
3-4. Graphical determination of eigenvalues in single isotropic bead formulation...113
3-5. Basis functions in single isotropic bead formulation...114
3-6. Diffusion of rod-coils compared to single isotropic bead formulation ... 115
4-1. Normalized diffusion of entangled coil-rod-coils in the small rod regime ... 121
4-4. Diffusion of entangled coil-rod-coils in the large rod regime ... 127
5-1. Sequence and electrophoresis of polypeptide genes and proteins...136
5-2. Synthesis of dye-labeled PEO-b-PA coil-rod-coil triblock copolymers ... 138
5-3. SDS-PAGE of anion exchange chromatography fractions from PEO-B3 reaction...139
5-4. Circular dichroism spectra of polypeptides...143
5-5. Small-angle neutron scattering of tracers in deuterated PEO solutions ... 145
5-6. Sam ple FR S results...147
5-7. D iffusion of PEO homopolymers...148
5-8. Diffusion of coil-rod-coil triblock copolymers with 40kDa coil blocks ... 149
5-9. Diffusion of triblock copolymers with coil block size varied ... 150
5-10. Comparison of experimental and simulation results ... 150
6-1. Synthesis of dye-labeled PEO-b-PA rod-coil diblock copolymers ... 161
6-2. Comparison of diblocks and triblocks in large rod regime by simulation...164
6-3. Corrected arm retraction mechanism in triblock dynamics...167
6-4. Comparison of diblocks and triblocks in large rod regime by experiments...168
6-5. Symmetry of rod-coil diblock copolymer reptation ... 171
6-6. Simulation and experimental results for diblocks and triblocks in small rod regime ... 173
6-7. Rotational correlation function of rods in diblocks and triblocks ... 174
7-1. Synthesis and activation of PPV-b-DTE-PI ... 184
7-2. Gel permeation chromatography of PPV-b-PI synthesis and irradiation ... 189
7-3. Sam ple FRS results of PPV -b-PI...190
7-4. Self-diffusion of polyisoprenes vs. molecular weight...192
7-5. Self-diffusion of PI and PPV-b-PI vs inverse temperature...193
7-6. Normalized self-diffusion of disordered isotropic PPV-b-PI...194
7-7. Activated reptation with constraint release mechanism ... 195
7-8. Normalized tracer diffusion of PEO-b-PA in different matrix molecular weights ... 199
7-9. Normalized tracer diffusion of PEO-b-PA vs. matrix molecular weight ... 199
8-1. Matching MD and slip-link for rod-coil block copolymers ... 211
8-2. Distribution of linear rod-coil positions along entanglement tube ... 212
8-3. Free energy and tube axis curvature of rod-coil rings along entanglement tube...214
8-4. Normalized curvilinear diffusion vs. rod length...216
9-1. Slip-link density and extension over time ... 226
9-2. Curvilinear motion of coil homopolymers ... 227
9-3. Normalized curvilinear diffusion of coil-rod-coil triblocks in the small rod regime ... 229
9-4. Effect of activated reptation on curvilinear diffusion...231
9-5. Curvilinear diffusion in the intermediate rod regime ... 233
9-6. R eptation path algorithm ... 235
9-7. Comparing reptation path with connected anchor tube definition ... 238
9-8. Effect of rod geometry on reptation path...239
9-9. Orientational correlation of reptation path ... 241
9-10. 3-dimensional center-of-mass diffusion of coil-rod-coil triblocks...243
A-1. Mean-squared displacement of entangled coil-rod-coil triblocks ... 256
A-2. Log-log mean-squared displacement of triblocks...256
A-3. Distribution of curvature mismatch energies...257
B-1. Gel permeation chromatography of PEO-B3-PEO ... 264
B-3. Form factors of tracer polym ers...266
B-4. Tracer diffusion of poly(ethylene oxide) homopolymers ... 267
C-1. Raw tracer diffusion of rod-coil diblock copolymers...268
D-1. Absorbance and emission spectra of PPV with DTE before and after UV irradiation...275
D-2. Small angle X-ray scattering of PPV-b-PI block copolymers...277
D-3. Order-disorder transition temperature of PPV-b-PI from SAXS ... 277
D-4. Birefringence intensity of PPV-b-PI block copolymers ... 278
D-5. Raw tracer diffusion of PEO-b-PA in aqueous PEG132k solutions...279
E-1. Matching MD and slip-link for coil homopolymers ... 280
E-2. Matching MD and slip-link for mean-squared displacement of rod homopolymers...282
E-3. Matching MD and slip-link for rotational correlation of rod homopolymers...283
E-4. Distribution of squared distance between adjacent anchor points for rods and coils ... 284
E-5. Local tube statistics of linear and ring polymers ... 28.5 E-6. Additional distributions of linear rod-coil positions along entanglement tube...286
E-7. Probability distributions of ring polymer positions along entanglement tube ... 286
E-8. Tube axis of ring polym ers ... 287
E-9. Additional free energy and tube axis curvatures of rod-coil rings...289
F-1. Orientational correlation of reptation path of coil homopolymers...292
F-2. Effect of A parameter for coil homopolymers on the reptation path definition ... 293
F-3. W orm like tube m odel schem atic...293
F-4. Center-of-mass diffusion of wormlike tube from simulation...295
List of Schemes
2-1. Synthesis of poly(alkoxyphenylene vinylene)... 39
2-2. Synthesis of PPV-b-PI... 45
2-3. Photoisomerization reaction of o-nitrostilbene... 61
2-4. Synthesis of maleimide-functionalized o-nitrostilbene ... 61
2-5. Photoisomerization reaction of dithienylethene ... 67
2-6. Synthesis of vinyl-functionalized dithienylethene ... 70
B-1. Synthesis of ONS-M ... 260
D-1. Synthesis of vinyl-DTE ... 270
D-2. Synthesis of poly(phenylene vinylene)...273
List of Tables
2-1. Parts list for FRS experiment... 555-1. Coil homopolymer and coil-rod-coil triblock copolymer tracers...140
5-2. Fractional helicities of polypeptides determined by circular dichroism...143
6-1. Rod-coil diblock copolymer tracers ... 161
Chapter 1: Background
1.1 Introduction
Rod-coil block copolymers1
-3 provide an elegant route to introduce functional rodlike
molecules into nanostructured polymer systems, including semiconducting polymers,3-5 rodlike
biomolecules such as polypeptides6,7 and polysaccharides,8~10 and engineering polymers such as polyimines and polyaramides."-14 These new functional polymers promise to advance
technology in areas such as polymer photovoltaics3, 15, 16 and LEDs,17-20 drug delivery devices,2 1 -23 tissue engineering gels,2 4
-26 polymer compatiblizers,5
and high strength polymer blends or composites. 1-14 For example, organic electronics have attracted a great deal of attention as third-generation photovoltaic (PV) materials due to the promise to dramatically decrease the cost of a PV module and for the ability to enable solar energy conversion in new forms such as paint and window shades. Producing high efficiency materials2 7-32 requires the formation of bulk
heterojunction nanostructures to efficiently dissociate excitons into free charge carriers to
generate power. Rod-coil block copolymers are increasingly investigated as a method to access the length scale of these bulk heterojunctions by self-assembly of thermodynamic equilibrium
nanostructures.3,16,17,32,33
In addition to these compelling technological applications, rod-coil block copolymers are also part of a wider class of hybrid functional materials, where individual domains are combined into shape amphiphiles with each motif contributing a different functionality along with an associated shape. Soft matter and polymer physics have recently devoted increasing attention to self-assembled systems composed of complex shapes,34' and advanced technological
applications have combined the physics of traditional Gaussian chain polymers with these rigid functional domains. PEGylated pharmaceuticals maintain the original fold and function of proteins while flexible polymers shield the molecule from enzymatic degradation.36 Hydrogels
for tissue engineering combine rigid associating domains with flexible linkers to produce physically assembled gels with unique mechanical properties.3 7 Protein-polymer conjugates3 8 are
actively explored for nanopatterning enzymes into catalytically active materials.39' 40 Polymer
nanocomposites have been demonstrated for high-performance engineering materials, where electronic, optical, and mechanical properties are independently tunable.4143 Therefore, the lessons learned by studying the self-assembly and dynamics of rod-coil block copolymers have a wide implications for a large class of materials.
Although a great deal of effort has been dedicated towards understanding the thermodynamics of rod-coil self-assembly for these many application areas, the melt or solution processing, diffusion, and self-assembly kinetics in these materials has barely been studied. Because all of these important dynamic properties depend on the same fundamental molecular mechanisms of polymer relaxation, the lack of fundamental understanding of dynamics represents a critical knowledge gap in the application of organic electronics, protein-based biomaterials, and rod/coil molecular composites. Dynamics of rod-coil block copolymers in the entangled regime is a particularly interesting academic problem because the nature of dynamic entanglement is fundamentally different between rods and coils. For entangled homopolymers, this difference has been shown to create divergent scaling behaviors for entangled rods44' 41
(diffusivity D ~ M, relaxation time rr A~?) and coils45-47 (D ~ M2
.3,
Tr M3 4). This thesis develops a fundamental theory for the dynamics of these molecules that accounts for the new physics that directly arises from the motion of multiple domains of dissimilar geometries within
the same molecule. Filling this crucial knowledge gap will enable engineers to rationally design processing strategies for the fabrication of organic electronic materials using molecular-level theories to understand mechanics, fluid dynamics, and diffusion processes.
The remainder of this chapter reviews the relevant background for this thesis, specifically the self-assembly of rod-coil block copolymers, established theories of polymer dynamics, and experimental and simulation techniques for testing these theories. It also provides an overview of the following thesis chapters.
1.2 Rod-Coil Block Copolymers
In comparison to well-known coil-coil diblock copolymers, the thermodynamic self-assembly of rod-coil systems is significantly more complex because both anisotropic liquid crystalline interactions and non-Gaussian chain topologies that result in significant changes in their phase behavior. Landau theories,4 8' 49 analytical scaling theories,50~54 self-consistent field theory,55-5 8 and coarse-grained molecular simulations59, 60 have all been applied to provide a theoretical and computational understanding of the thermodynamics behind self-assembly. These approaches indicate that in addition to the coil fraction,
#,
and Flory-Huggins interaction, XN, that are traditionally used to characterize assembly in a coil-coil diblock copolymer, self-assembly in rod-coil systems is also characterized by the dimensional asymmetry between rod and coil, , and the Maier-Saupe liquid crystalline interaction, pN. Experimental phase diagrams have been developed for both polymeric58' 61-65 and oligomeric66-70 rod-coil block copolymers,between the rod and coil. In addition, both microphase separation and liquid crystalline ordering are observed. A number of phases have been reported in rod-coil block copolymers that are not seen in coil-coil systems, including rectangular strip or puck phases,66,71, 72 wavy lamellar and
zig-zag phases,7 3
,7 4 and liquid crystalline hexagonally perforated lamellar phases.64 Experimental methods have been developed for the independent measurement of both the Maier-Saupe parameter7 5 characterizing liquid crystalline interactions and the Flory-Huggins interaction58
measuring repulsive interactions between the rod and coil. Using the temperature dependence of these parameters allows the experimental phase diagrams to be universalized,58 providing a direct, quantitative comparison with theories. Although quantitative agreement between theory and experiment has not been achieved with current modeling efforts, the comparison confirms most of the qualitative features of the models.
Despite extensive research on the self-assembly of rod-coil systems, little is known about the dynamics of these molecules. A knowledge of dynamics is central to understanding both the kinetics of self-assembly and the flow properties of rod-coil systems for the development of industrial processes employing these new materials. For example, it is hypothesized that kinetic effects on material self-assembly contribute to the formation of zig-zag and wavy lamellar structures in some rod-coil block copolymers.73' 74 Understanding the dynamics of rod-coil polymers will provide insight into how and why these phases form. In addition, the rate of structure formation during annealing processes and the mechanical properties76 of the resulting self-assembled structures will depend strongly on the dynamic properties of the block copolymers. These dynamic phenomena have only been explored in a few isolated studies. Rheological measurements have been used to identify order-disorder transitions77' 78 and to
measure intrinsic viscosities.79 Borsali, et al. also provided analytical expressions for dynamic structure factors in dilute solution.80
1.3 Theories of Polymer Dynamics
The study of polymer dynamics has developed fundamental theories that have successfully predicted many properties such as diffusion, rheology, and scattering phenomena. Theories of polymer dynamics can apply to enormous libraries of different molecules from DNA81, 82 to
commodity polymers such as polystyrene plastics83 or polyisoprene rubbers,84 because many dynamic properties depend on length scales larger than the individual chemical species. At these length scales, polymers can be universalized into common motifs such as a Gaussian coil, where successive chain monomers are arranged according to a random walk, or a rigid rod. The dynamics can then be described by scaling relationships between universal parameters such as temperature, concentration, molecular weight, etc.
One of the simplest theories of polymer dynamics is the Rouse model, which describes the motion of flexible polymers in dilute solution or unentangled melts.85 Polymers are modeled as a series of non-interacting beads connected by Hookean springs undergoing Brownian motion in the Stokes limit. At long time scales, the Rouse model effectively captures localized interactions along the polymer chain while neglecting long-range effects such as hydrodynamic or excluded volume interactions. The motion of monomers is governed by non-inertial Langevin equations that can be solved analytically by diagonalization and expression in terms of normal modes.45 Some key predictions include diffusion D ~M', viscosity / ~ M1, and relaxation time r ~ M2for
molecular weight M, which are quantitatively valid for unentangled melts where interactions between monomers are screened. Predictions of the Rouse model provide a convenient theoretical framework and a benchmark for comparison with more complex systems. For
example, the Zimm model is a related theory that includes hydrodynamic interactions, with slightly different predictions that are more applicable for polymers in dilute solution.86
When polymers are sufficiently long or concentrated, physical entanglements between chains cause a change in the molecular mechanism of diffusion. Above a phenomenological size known as the entanglement molecular weight Me, topological interactions that prohibit chains from crossing each other have a large effect on dynamics. The dynamics of entangled polymers have been successfully described by reptation theories, which consider the motion of a single tracer polymer in an effective matrix of surrounding chains.45' 47' 87 To preserve topological integrity, the matrix is replaced with an effective tube of diameter a (Figure 1-1), and the tracer can freely diffuse along the tube's contour but is prohibited from moving perpendicular to it. The centerline of the tube is called the primitive path. As the tube is merely a mean-field interpretation of the matrix, it is constantly created and destroyed as the tracer diffuses along its contour. The lifetime of the tube is governed by the presence of the matrix chains, and the parameter M depends on characteristics of the matrix, such as its concentration and chemical details.
Application of the reptation model to coil polymers leads to predictions of dynamical phenomena that significantly depart from the unentangled Rouse model. Since the coil's tube is a random walk of step size a, scaling analysis predicts that the longest relaxation time is r - 113,
after which the rotation of the molecule is unhindered and the polymer diffuses a distance of order Rg. This leads directly to predictions for diffusivity D ~ M 2 and viscosity
7 ~ M3.
Experimental studies of tracer and self-diffusion have consistently produced scaling exponents between -2 and -2.5.46,88-93 This discrepancy is explained by more advanced theories that account for the motion of the surrounding chains94-96 and fluctuations in the length of the tube.97'98 These
predict that diffusivity scales as D M2 3 and viscosity scales as q ~ M34, in line with
experiment. The first effect is known as constraint release, where the tube is renewed over time
as the surrounding entanglements relax. These effects are commonly studied using blends of short and long linear polymers,99' 100 where the short polymers act as transient entanglements for the long chains. The second effect is called contour length fluctuation, where the flexibility of the polymer allows the ends to retract into the tube and explore other regions in space. These effects are commonly studied using star polymers,10
1-104 where contour length fluctuation (also known as arm retraction) is the only viable relaxation mechanism.
The motion of rigid rod polymers is qualitatively different from flexible coils. Unentangled rods move according to an anisotropic mobility tensor, whose translational components scale according to L-1 and rotational diffusivity follows DrO ~ L- , where L is rod length. Hydrodynamic interactions add minor corrections to these expressions.4 5 For rod polymers in the entangled regime, analogous reptation theories pioneered by Doi and Edwards4 4
, 45, 105 also predict that
translational diffusion parallel to the entanglement tube is essentially unhindered while perpendicular diffusion is forbidden.106 This results in a D - L-1 scaling identical to a dilute
solution. Experimental measurements show a D ~ L-.8 scaling, which is explained by higher order effects such as flexibility and hydrodynamic interactions.107' 108 Rod polymers differ
substantially from coil polymers in rotational diffusion, because the large spatial extent of the rod compared to the entanglement tube radius means even complete diffusion out of the tube only allows small rotational relaxations. The Doi-Edwards model predicts Dr ~ Dro(a/L)2 , so
Dr - L-5 for tracer rods diffusing through another matrix where a is fixed. For self-diffusion, the dependence of tube diameter on rod length a ~ L-2 can be derived from geometrical arguments, so the theoretical prediction is Dr ~ L-9, which has been confirmed by experiment and simulation. 109-111
While the theories of entangled dynamics of rod and coil polymers have been well-developed, there has been no systematic effort to describe the relaxation mechanisms of rod-coil block copolymers to our knowledge. This thesis applies reptation theories to these polymers, highlighting the new physics that arises when the two domains of very different flexibility and geometry are combined onto the same molecule.
1.4 Measurements by Experiments and Simulation
The theories of polymer dynamics are tested in experiments and simulation by measuring dynamical properties and comparing with predictions. For example, diffusion measurements describe the behavior of systems at long time scales and close to equilibrium.112, 113 Rheology
and dielectric spectroscopy can span a long range of time scales and describe behavior both close and far from equilibrium using linear and nonlinear deformations.47' 104 Inelastic neutron
scattering can measure dynamic scattering functions at very short time scales.1 14 A successful theory of polymer dynamics must correctly explain all of these observables before it can be applied to real engineering problems. As a first theoretical treatment of entangled rod-coil block copolymers, this thesis focuses on predicting diffusion phenomena.
Experimentally, diffusion measurements are most commonly performed by generating a concentration gradient, monitoring the rate of its evolution, and extracting a diffusivity by comparing the data to a solution of Fick's second law. Since diffusion in polymer melts is easily slower than 10-2 m2/s, the length scale of the concentration gradients must be sufficiently small so that their evolution can be observed over experimental time scales. Ex situ methods such as forward recoil spectrometry (FRES), Rutherford backscattering (RBS), secondary ion mass spectrometry (SIMS), and neutron reflectivity (NR) are commonly used for very slow processes
(< 10-19 m2/s).112 In these procedures, thin (-100 nm) layers of isotopically labeled polymers are brought together and allowed to diffuse for a specified amount of time before diffusion is quenched by lowering the temperature. The resulting concentration profile is measured by depth profiling using a variety of techniques. Faster diffusivities can be measured by generating a concentration perturbation and then measuring their evolution in situ. In pulsed-field gradient nuclear magnetic resonance (PFG-NMR), diffusion is extracted from the precession of nuclear spins. This technique is attractive because it does not require fluorescent or isotopic labels, but the accessible diffusivities are typically above 10-14 m2/s." 5 In fluorescence recovery after
photobleaching (FRAP), molecules are labeled with a photobleachable fluorescent dye, a pattern is bleached into a sample, and the evolution of this pattern is observed under a microscope.1 16 Since the pattern must be optically resolvable, the range of diffusivities is similar to PFG-NMR. Forced Rayleigh scattering (FRS) is similar to FRAP, where a photochromic dye is attached to
the polymer and a pattern is written into the sample.1 13
, 117, 118 However, this pattern is written by
holographic interference of coherent laser beams where the length scale can be smaller, so the accessible range is 104--10-19 m2/s. FRS is the diffusion measurement technique that is used throughout this thesis, and it is explained extensively in the next chapter.
Entangled polymer dynamics can also be simulated using molecular dynamics (MD) models. Since entanglement phenomena are applicable to a library of chemical species, models are typically coarse-grained above the level of atomistic detail. In the most common model developed by Kremer and Grest,119 polymers are modeled as a series of monomers connected by springs, with excluded volume interactions between all monomers that are tuned to prevent chains from crossing each other. This model is used extensively throughout this thesis and explained in detail in subsequent chapters. Beyond the dynamical properties such as diffusion from mean-squared displacement or other correlation functions, simulations can test the theories with levels of detail that are difficult to attain in experiments. For example, the mean-squared displacement of individual monomers or the rotational diffusion are easily calculated from molecular dynamics, but require expensive neutron scattering20
, 121 with sophisticated labeling techniques9" or complex dichroism measurements22 in experiments. Recently, algorithms have
also been developed that enable visualization and quantification of the entangled tube,12 3-130 directly testing the reptation concepts. These have led to more coarse-grained models, where entanglements can be represented by simple interactions between chains called slip-links, rather than the collective topological effects of excluded volume between many monomers. 3 1-133 These
slip-link models can reveal deeper insights on the nature of entanglement dynamics with enormous computational speedups compared to MD, while matching data from experiments and detailed simulations.
1.5 Thesis Overview
This thesis describes a systematic study of the dynamics of entangled rod-coil block copolymers, developing a theory for the molecular mechanisms of motion for this class of polymers. Chapter 2 reviews details of the experiments and simulations that are used throughout
the work. Chapter 3 considers the motion of unentangled polymers by extending the Rouse model to rod-coils and solving the equations using Brownian dynamics simulation, showing that the long-time diffusion behavior is captured by the sum of the friction of the rod and the coil. Chapter 4 studies the entangled dynamics of symmetric coil-rod-coil triblock copolymers in a melt of coil homopolymers using the Kremer-Grest model for molecular dynamics simulation. It predicts that the diffusion of rod-coils is slower than both rod and coil homopolymers because of a mismatch in the natural curvature of the entanglement tubes of these two domains, and it proposes relaxation mechanisms known as activated reptation and arm retraction that govern regimes of short and long rods. These predictions are verified in Chapter 5 using experimental tracer diffusion measurements by forced Rayleigh scattering on copolymers with rods composed of alanine-rich polypeptides and coils composed of poly(ethylene oxide). In Chapter 6, these relaxation mechanisms are applied to the tracer diffusion of rod-coil diblock copolymers, and key questions regarding the asymmetry of these molecules are addressed using both experimental and simulation measurements. Chapter 7 contains experimental self-diffusion measurements of isotropic disordered rod-coil melts composed of poly(phenylene vinylene) rods and polyisoprene coils, and the proposed relaxation mechanisms are refined to include the motion of the surrounding entanglements. Chapter 8 develops a coarse-grained slip-link model to present direct
definitive evidence of the activated reptation mechanism. Finally, Chapter 9 applies the slip-link model to explore the competition between these two mechanisms in the intermediate rod regime.
1.6 References
1. Lee, M.; Cho, B. K.; Zin, W. C. Chem. Rev. 2001, 101, (12), 3869-3892. 2. Olsen, B. D.; Segalman, R. A. Mat. Sci. Eng. R 2008, 62, (2), 37-66.
3. Segalman, R. A.; McCulloch, B.; Kirmayer, S.; Urban, J. J. Macromolecules 2009, 42, (23), 9205-9216.
4. de Boer, B.; Stalmach, U.; van Hutten, P. F.; Melzer, C.; Krasnikov, V. V.; Hadziioannou, G. Polymer 2001, 42, (21), 9097-9109.
5. Sivula, K.; Ball, Z. T.; Watanabe, N.; Frechet, J. M. J. Advanced Materials 2006, 18, (2), 206.
6. van Hest, J. C. M. Polym. Rev. 2007, 47, (1), 63-92.
7. Klok, H. A. J. Polym. Sci. A. Polym. Chem. 2005, 43, (1), 1-17. 8. Loos, K.; Muller, A. H. E. Biomacromolecules 2002, 3, (2), 368-373. 9. Haddleton, D. M.; Ohno, K. Biomacromolecules 2000, 1, (2), 152-156.
10. Akiyoshi, K.; Kohara, M.; Ito, K.; Kitamura, S.; Sunamoto, J. Macromolecular Rapid
Communications 1999, 20, (3), 112-115.
11. Konig, H. M.; Gorelik, T.; Kolb, U.; Kilbinger, A. F. M. Journal of the American
Chemical Society 2007, 129, (3), 704-708.
12. Pae, Y.; Harris, F. W. Journal of Polymer Science Part a-Polymer Chemistry 2000, 38, (23), 4247-4257.
13. Harris, F. W.; Livengood, B. P.; Ding, H.; Lin, F. L.; Cheng, S. Z. D. Thermochimica
Acta 1996, 272, 157-169.
14. Takayanagi, M.; Ogata, T.; Morikawa, M.; Kai, T. Journal of Macromolecular
Science-Physics 1980, B17, (4), 591-615.
15. Yu, G.; Gao, J.; Hummelen, J. C.; Wudl, F.; Heeger, A. J. Science 1995, 270, (5243), 1789-1791.
16. Tao, Y. F.; McCulloch, B.; Kim, S.; Segalman, R. A. Soft Matter 2009, 5, (21), 4219-4230.
17. Chochos, C. L.; Kallitsis, J. K.; Gregoriou, V. G. Journal of Physical Chemistry B 2005, 109, (18), 8755-8760.
18. Becker, S.; Ego, C.; Grimsdale, A. C.; List, E. J. W.; Marsitzky, D.; Pogantsch, A.; Setayesh, S.; Leising, G.; Mullen, K. Synthetic Metals 2001, 125, (1), 73-80.
19. Osaheni, J. A.; Jenekhe, S. A. Journal of the American Chemical Society 1995, 117, (28), 7389-7398.
20. Tao, Y. F.; Ma, B. W.; Segalman, R. A. Macromolecules 2008, 41, (19), 7152-7159. 21. Bellomo, E. G.; Wyrsta, M. D.; Pakstis, L.; Pochan, D. J.; Deming, T. J. Nature
22. Checot, F.; Lecommandoux, S.; Gnanou, Y.; Klok, H. A. Angewandte
Chemie-International Edition 2002, 41, (8), 1339-1343.
23. Vandermeulen, G. W. M.; Tziatzios, C.; Klok, H. A. Macromolecules 2003, 36, (11), 4107-4114.
24. Breedveld, V.; Nowak, A. P.; Sato, J.; Deming, T. J.; Pine, D. J. Macromolecules 2004, 37, (10), 3943-3953.
25. Nowak, A. P.; Breedveld, V.; Pakstis, L.; Ozbas, B.; Pine, D. J.; Pochan, D.; Deming, T.
J. Nature 2002, 417, (6887), 424-428.
26. Nagapudi, K.; Brinkman, W. T.; Leisen, J.; Thomas, B. S.; Wright, E. R.; Haller, C.; Wu, X. Y.; Apkarian, R. P.; Conticello, V. P.; Chaikof, E. L. Macromolecules 2005, 38, (2), 345-354. 27. Blom, P. W. M.; Mihailetchi, V. D.; Koster, L. J. A.; Markov, D. E. Advanced Materials 2007, 19, (12), 1551-1566.
28. Coakley, K. M.; McGehee, M. D. Chemistry ofMaterials 2004, 16, (23), 4533-4542. 29. Hoppe, H.; Sariciftci, N. S. Journal of Materials Research 2004, 19, (7), 1924-1945. 30. Jorgensen, M.; Norrman, K.; Krebs, F. C. Solar Energy Materials and Solar Cells 2008, 92, (7), 686-714.
31. Po, R.; Maggini, M.; Camaioni, N. Journal of Physical Chemistry C 2010, 114, (2), 695-706.
32. Yang, X.; Loos, J. Macromolecules 2007, 40, (5), 1353-1362.
33. Boudouris, B. W.; Molins, F.; Blank, D. A.; Frisbie, C. D.; Hillmyer, M. A.
Macromolecules 2009, 42, (12), 4118-4126.
34. Glotzer, S. C.; Solomon, M. J. Nature Mater. 2007, 6, (8), 557-562.
35. Sacanna, S.; Korpics, M.; Rodriguez, K.; Colon-Melendez, L.; Kim, S. H.; Pine, D. J.; Yi, G. R. Nature Communications 2013, 4.
36. Harris, J. M.; Chess, R. B. Nature Reviews Drug Discovery 2003, 2, (3), 214-22 1.
37. Shen, W.; Zhang, K. C.; Korfield, J. A.; Tirrell, D. A. Nature Mater. 2006, 5, (2),
153-158.
38. Borner, H. G. Progress in Polymer Science 2009, 34, (9), 811-851.
39. Thomas, C. S.; Glassman, M. J.; Olsen, B. D. ACSNano 2011, 5, (7), 5697-5707. 40. Thomas, C. S.; Xu, L. Z.; Olsen, B. D. Biomacromolecules 2012, 13, (9), 2781-2792. 41. Winey, K. I.; Vaia, R. A. Mrs Bulletin 2007, 32, (4), 314-319.
42. Bockstaller, M. R.; Mickiewicz, R. A.; Thomas, E. L. Advanced Materials 2005, 17, (11), 1331-1349.
43. Balazs, A. C.; Emrick, T.; Russell, T. P. Science 2006, 314, (5802), 1107-1110. 44. Doi, M.; Edwards, S. F. J. Chem. Soc., Faraday Trans. 111978, 74, 560-570.
45. Doi, M.; Edwards, S. F., The Theory ofPolymer Dynamics. Clarendon: Oxford, 1986. 46. Tao, H.; Lodge, T. P.; von Meerwall, E. D. Macromolecules 2000, 33, (5), 1747-175 8. 47. McLeish, T. C. B. Adv. Phys. 2002, 51, (6), 1379-1527.
48. Holyst, R.; Schick, M. Journal of Chemical Physics 1992, 96, (1), 730-740. 49. Hammouda, B. J. Chem. Phys. 1993, 98, (4), 3439-3444.
50. Semenov, A. N. Molecular Crystals and Liquid Crystals 1991, 209, 191-199.
51. Semenov, A. N.; Vasilenko, S. V. Zhurnal Eksperimentalnoi I Teoreticheskoi Fiziki 1986, 90, (1), 124-140.
52. Halperin, A. Europhysics Letters 1989, 10, (6), 549-553. 53. Halperin, A. Macromolecules 1990, 23, (10), 2724-273 1.
55. Matsen, M. W.; Barrett, C. Journal of Chemical Physics 1998, 109, (10), 4108-4118.
56. Pryamitsyn, V.; Ganesan, V. J. Chem. Phys. 2004, 120, (12), 5824-5838.
57. Tao, Y. F.; Olsen, B. D.; Ganesan, V.; Segalman, R. A. Macromolecules 2007, 40, (9), 3320-3327.
58. Olsen, B. D.; Shah, M.; Ganesan, V.; Segalman, R. A. Macromolecules 2008, 41, (18), 6809-6817.
59. Horsch, M. A.; Zhang, Z. L.; Glotzer, S. C. Physical Review Letters 2005, 95, (5). 60. Horsch, M. A.; Zhang, Z. L.; Glotzer, S. C. Journal of Chemical Physics 2006, 125, (18). 61. Olsen, B. D.; Segalman, R. A. Macromolecules 2005, 38, (24), 10127-10137.
62. Olsen, B. D.; Segalman, R. A. Macromolecules 2006, 39, (20), 7078-7083. 63. Olsen, B. D.; Segalman, R. A. Macromolecules 2007, 40, (19), 6922-6929.
64. Tenneti, K. K.; Chen, X. F.; Li, C. Y.; Tu, Y. F.; Wan, X. H.; Zhou,
Q.
F.; Sics, I.; Hsiao, B. S. Journal of the American Chemical Society 2005, 127, (44), 15481-15490.65. Ho, V.; Boudouris, B. W.; McCulloch, B. L.; Shuttle, C. G.; Burkhardt, M.; Chabinyc, M. L.; Segalman, R. A. Journal of the American Chemical Society 2011, 133, (24), 9270-9273. 66. Radzilowski, L. H.; Carragher, B. 0.; Stupp, S. I. Macromolecules 1997, 30, (7), 2110-2119.
67. Cho, B. K.; Lee, M.; Oh, N. K.; Zin, W. C. Journal of the American Chemical Society 2001, 123, (39), 9677-9678.
68. Lee, M.; Oh, N. K.; Lee, H. K.; Zin, W. C. Macromolecules 1996, 29, (17), 5567-5573. 69. Lee, M.; Cho, B. K.; Kim, H.; Yoon, J. Y.; Zin, W. C. Journal ofthe American Chemical
Society 1998, 120, (36), 9168-9179.
70. Lee, M.; Cho, B. K.; Kim, H.; Zin, W. C. Angewandte Chemie-International Edition 1998, 37, (5), 638-640.
71. Radzilowski, L. H.; Wu, J. L.; Stupp, S. I. Macromolecules 1993, 26, (4), 879-882. 72. Radzilowski, L. H.; Stupp, S. I. Macromolecules 1994, 27, (26), 7747-7753.
73. Chen, J. T.; Thomas, E. L.; Ober, C. K.; Mao, G. P. Science 1996, 273, (5273), 343-346. 74. Chen, J. T.; Thomas, E. L.; Ober, C. K.; Hwang, S. S. Macromolecules 1995, 28, (5), 1688-1697.
75. Olsen, B. D.; Jang, S. Y.; Luning, J. M.; Segalman, R. A. Macromolecules 2006, 39, (13), 4469-4479.
76. Muller, C.; Goffri, S.; Breiby, D. W.; Andreasen, J. W.; Chanzy, H. D.; Janssen, R. A. J.; Nielsen, M. M.; Radano, C. P.; Sirringhaus, H.; Smith, P.; Stingelin-Stutzmann, N. Advanced
Functional Materials 2007, 17, (15), 2674-2679.
77. Gopalan, P.; Zhang, Y. M.; Li, X. F.; Wiesner, U.; Ober, C. K. Macromolecules 2003, 36, (9), 3357-3364.
78. Olsen, B. D.; Teclemariam, N. P.; Muller, S. J.; Segalman, R. A. Soft Matter 2009, 5, (12), 2453-2462.
79. Yoda, R.; Hirokawa, Y.; Hayashi, T. Eur. Polym. J. 1994, 30, (12), 1397-1401.
80. Borsali, R.; Lecommandoux, S.; Pecora, R.; Benoit, H. Macromolecules 2001, 34, (12), 4229-4234.
81. Perkins, T. T.; Smith, D. E.; Chu, S. Science 1994, 264, (5160), 819-822. 82. Duke, T.; Viovy, J. L. Phys. Rev. E 1994, 49, (3), 2408-2416.
83. Amis, E. J.; Han, C. C.; Matsushita, Y. Polymer 1984, 25, (5), 650-658. 84. Fleischer, G.; Appel, M. Macromolecules 1995, 28, (21), 7281-7283. 85. Rouse, P. E. J. Chem. Phys. 1953, 21, (7), 1272-1280.
86. Zimm, B. H. J. Chem. Phys. 1956, 24, 269.
87. Degennes, P. G. Journal of Chemical Physics 1971, 55, (2), 572-&.
88. Antonietti, M.; Coutandin, J.; Grutter, R.; Sillescu, H. Macromolecules 1984, 17, (4), 798-802.
89. Leger, L.; Hervet, H.; Rondelez, F. Macromolecules 1981, 14, (6), 1732-1738.
90. Kim, H. D.; Chang, T. Y.; Yohanan, J. M.; Wang, L.; Yu, H. Macromolecules 1986, 19, (11), 2737-2744.
91. Wesson, J. A.; Noh, I.; Kitano, T.; Yu, H. Macromolecules 1984, 17, (4), 782-792. 92. Wheeler, L. M.; Lodge, T. P. Macromolecules 1989, 22, (8), 3399-3408.
93. Nemoto, N.; Kishine, M.; Inoue, T.; Osaki, K. Macromolecules 1991, 24, (7), 1648-1654. 94. Wang, Z. W.; Larson, R. G. Macromolecules 2008, 41, (13), 4945-4960.
95. Zamponi, M.; Wischnewski, A.; Monkenbusch, M.; Willner, L.; Richter, D.; Likhtman, A. E.; Kali, G.; Farago, B. Phys. Rev. Lett. 2006, 96, (23).
96. de Gennes, P. G. Macromolecules 1976, 9, (4), 587-593.
97. Milner, S. T.; McLeish, T. C. B. Phys. Rev. Lett. 1998, 81, (3), 725-728.
98. Zamponi, M.; Monkenbusch, M.; Willner, L.; Wischnewski, A.; Farago, B.; Richter, D.
Europhysics Letters 2005, 72, (6), 1039-1044.
99. Doi, M.; Graessley, W. W.; Helfand, E.; Pearson, D. S. Macromolecules 1987, 20, (8), 1900-1906.
100. Viovy, J. L.; Rubinstein, M.; Colby, R. H. Macromolecules 1991, 24, (12), 3587-3596. 101. Doi, M.; Kuzuu, N. Y. J. Polym. Sci. C 1980, 18, (12), 775-780.
102. Milner, S. T.; McLeish, T. C. B. Macromolecules 1997, 30, (7), 2159-2166. 103. Pearson, D. S.; Helfand, E. Macromolecules 1984, 17, (4), 888-895.
104. Watanabe, H. Progress in Polymer Science 1999, 24, (9), 1253-1403.
105. Doi, M.; Edwards, S. F. J. Chem. Soc., Faraday Trans. 111978, 74, 918-932. 106. Ramanathan, S.; Morse, D. C. Phys. Rev. E 2007, 76, (1), -.
107. Tracy, M. A.; Pecora, R. Annu. Rev. Phys. Chem. 1992, 43, 525-557. 108. Odijk, T. Macromolecules 1983, 16, (8), 1340-1344.
109. Maguire, J. F.; McTague, J. P.; Rondelez, F. Phys. Rev. Lett. 1980, 45, (23), 1891-1894. 110. Cobb, P. D.; Butler, J. E. J. Chem. Phys. 2005, 123, (5).
111. Phalakornkul, J. K.; Gast, A. P.; Pecora, R. Macromolecules 1999, 32, (9), 3122-3135. 112. Yokoyama, H. Mat. Sci. Eng. R 2006, 53, (5-6), 199-248.
113. Lodge, T.; Chapman, B. Trends Polym. Sci. 1997, 5, (4), 122-128.
114. Richter, D.; Monkenbusch, M.; Arbe, A.; Colmenero, J. Neutron Spin Echo in Polymer
Systems 2005, 174, 1-221.
115. Price, W. S. Concepts in Magnetic Resonance 1997, 9, (5), 299-336.
116. Axelrod, D.; Koppel, D. E.; Schlessinger, J.; Elson, E.; Webb, W. W. Biophysical
Journal 1976, 16, (9), 1055-1069.
117. Sillescu, H.; Ehlich, D., Applications of Holographic Grating Techniques to the Study of Diffusion Processes in Polymers. In Lasers in Polymer Science and Technology: Applications, Fouassier, J. P.; Rabek, J. F., Eds. CRC Press: Boca Raton, FL, 1990; Vol. 3, pp 211-226.
118. Schartl, W., Forced Rayleigh Scattering - Principles and Application (Self Diffusion of Spherical Nanoparticles and Copolymer Micelles). In Soft Matter Characterization, Borsali, R.; Pecora, R., Eds. Springer Netherlands: 2008; Vol. 1, pp 677-703.
120. Schleger, P.; Farago, B.; Lartigue, C.; Kollmar, A.; Richter, D. Phys. Rev. Lett. 1998, 81, (1), 124-127.
121. Wischnewski, A.; Monkenbusch, M.; Willner, L.; Richter, D.; Kali, G. Phys. Rev. Lett.
2003, 90, (5), -.
122. Hogan, M.; Dattagupta, N.; Crothers, D. M. Proc. Nati. Acad. Sci. U.S.A. 1978, 75, (1),
195-199.
123. Everaers, R.; Sukumaran, S. K.; Grest, G. S.; Svaneborg, C.; Sivasubramanian, A.; Kremer, K. Science 2004, 303, (5659), 823-826.
124. Kremer, K. Computer Simulations in Condensed Matter Systems: From Materials to
Chemical Biology, Vol 2 2006, 704, 341-378
598.
125. Sukumaran, S. K.; Grest, G. S.; Kremer, K.; Everaers, R. J. Polym. Sci. B 2005, 43, (8), 917-933.
126. Foteinopoulou, K.; Karayiannis, N. C.; Mavrantzas, V. G.; Kroger, M. Macromolecules 2006, 39, (12), 4207-4216.
127. Hoy, R. S.; Foteinopoulou, K.; Kroger, M. Phys. Rev. E 2009, 80, (3), 031803. 128. Larson, R. G. J. Polym. Sci. B 2007, 45, (24), 3240-3248.
129. Zhou, Q.; Larson, R. G. Macromolecules 2005, 38, (13), 5761-5765. 130. Likhtman, A. E. Soft Matter 2014, 10, (12), 1895-1904.
131. Likhtman, A. E. Macromolecules 2005, 38, (14), 6128-6139.
132. Sukumaran, S. K.; Likhtman, A. E. Macromolecules 2009, 42, (12), 4300-4309. 133. Nair, D. M.; Schieber, J. D. Macromolecules 2006, 39, (9), 3386-3397.
Chapter 2: Methods
2.1 Synthesis of Rod-Coil Block Copolymers
Two model systems of rod-coil block copolymers are used in this thesis. The first system is used to measure tracer diffusion of rod-coils through an entangled coil homopolymer matrix. Rod blocks are composed of alanine-rich a-helical polypeptides (PA), coil blocks are poly(ethylene oxide) (PEO), and the matrix is an aqueous PEO solution. In the second system, the self-diffusion of rod-coils is measured in the isotropic disordered state. Rod blocks are composed of poly(alkoxyphenylene vinylene) (PPV), and coil blocks are polyisoprene (PI). The synthesis of both systems is described in the next sections.
2.1.1 Synthesis of poly(ethylene glycol)-b-polyalanine copolymers
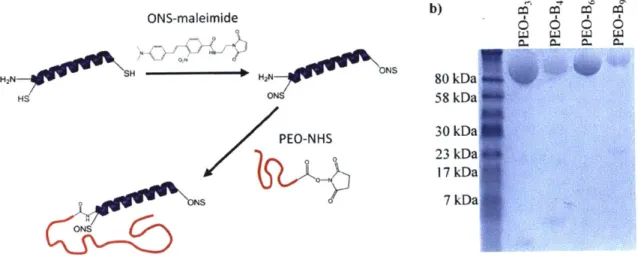
The synthesis of PEO-b-PA rod-coil block copolymers is summarized in Figure 2-1, and described below.
Recombinant construction of polypeptide genes. A gene encoding for an alanine-rich polypeptide was kindly provided by K. L. Kiick in the pET19b-RF1-B6 plasmid.1 This rodlike
polypeptide is a hexamer of the peptide sequence AAAQAAQAQAAAEAAAQAAQAQ, denoted as B throughout this work. This repeat unit has a length of 3.3 nm and a molar mass of 1982 Da. All restriction digests, PCR amplifications, and ligations were performed using enzymes and protocols from New England BioLabs (Ipswich, MA). Digests were followed by purification by agarose gel electrophoresis labeled by ethidium bromide, using cleanup kits from
Cloning Expression Metal-affinity purification
o6
PEO-NHS ONS-maleimide 0 0 --NH2 ONSH2N ONS ion-exchange ONS
ONs chromatography
Figure 2-1. Synthesis of dye-labeled poly(ethylene glycol)-b-polyalanine coil-rod-coil triblock copolymers.
Qiagen (Valencia, CA). PCR was followed by purification using either electrophoresis or desalting kits from Qiagen. Ligations were followed by transformation into chemically competent XL1Blue cells, prepared using a kit from Zymo Research (Irvine, CA). Site-directed mutagenesis was performed with a Phusion kit from Thermo (Pittsburgh, PA), with primer design as described in the included literature. Primers were obtained from IDT (Coralville, IA).
The gene encoding B6 was amplified directly from the pET19b-RFI-B6 plasmid by polymerase chain reaction (PCR) flanked by PstI and HindIII restriction sites,' and digested and ligated into the multiple cloning site of the pQE9 expression plasmid (Figure 2-2f). Genes encoding B3, B4, and B9 were constructed using the methods adapted from Farmer, et al.' An oligonucleotide encoding the B monomer was incorporated into the common pUC19 plasmid (Figure 2-2a), flanked by the type Ils restriction site BsaI. PCR amplification, restriction digest
by BsaI, and purification by agarose gel electrophoresis produced the 66 bp double stranded
DNA encoding the B sequence with identical GCAG overhangs at the 5' and 3' ends. A pET28a plasmid was mutated to incorporate a BsaI restriction site (Figure 2-2b) and digested to give the same GCAG overhang. The B gene was concatemerized into this plasmid (Figure 2-2c), by ligating the plasmid with the maximum amount of B insert in the ligation protocol. For example, a typical 20 pL reaction would include 3 pL containing 100 ng of plasmid, 2 gL of 1 Ox T4 ligase buffer, 14 p.L of B insert, and 1 p.L of T4 ligase, incubated for 1 hr at 16'C. To create the longer B9 oligomer, the concatemerization was repeated twice, first yielding B3 and then B3n. The genes encoding B. were then digested from pQE28a using PstI and HindIlI and ligated into the pQE9 expression plasmid (Figure 2-2d). A few single amino acid residues were then introduced by site-directed mutagenesis: a tryptophan near the N-terminus to increase the A280 signal, a lysine
near the C-terminus for polymer conjugation, and cysteines near the N- and C-termini for attachment of a photochromic dye. These mutations are shown in Figure 2-2e and Figure 2-3a. All final plasmids were confirmed by restriction analysis (Figure 2-3b) and dideoxy sequencing
(Genewiz, Cambridge, MA).
Bacterial expression of polypeptides. pQE9 expression plasmids containing a series of genes encoding for Bn oligomers of varying length were transformed into the SG13009(pREP4) expression host. A single bacterial colony was used to inoculate 5 mL of Lysogeny Broth with ampicillin (200 mg/L) and kanamycin (50 mg/L), which was shaken overnight at 37'C. This culture then inoculated 1 L of Terrific Broth with the same antibiotic concentrations and temperature. At OD60 0 = 0.8-1 which occurred after 3-4 hours, expression was induced with isopropyl