HAL Id: tel-01743708
https://tel.archives-ouvertes.fr/tel-01743708
Submitted on 26 Mar 2018HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Calcul efficace de la structure des protéines à partir de
contacts évolutifs
Fabrice Allain
To cite this version:
Fabrice Allain. Calcul efficace de la structure des protéines à partir de contacts évolutifs. Bio-Informatique, Biologie Systémique [q-bio.QM]. Université Pierre et Marie Curie - Paris VI, 2017. Français. �NNT : 2017PA066366�. �tel-01743708�
Université Pierre et Marie Curie
École doctorale Complexité du Vivant
Unité de Bioinformatique Structurale Institut Pasteur
Thèse de doctorat en Sciences de la Vie
Calcul efficace de la structure des protéines à
partir de contacts évolutifs
Fabrice Allain
Directeur de thèse : Pr. Michael Nilges
Co encadrant de thèse : Dr. Benjamin Bardiaux
Présentée et soutenue publiquement le 30 Novembre 2017 Jury
Pr. Martin Weigt (PU)
Président
Dr. Pierre Tufféry (DR)
Rapporteur
Dr. Jean-Christophe Gelly (MCU) Rapporteur
Dr. Sophie Zinn-Justin (CR)
Examinateur
Pr. Michael Nilges (PR)
Examinateur
Dr. Benjamin Bardiaux (CR)
Examinateur
Except where otherwise noted, this work is licensed under https://creativecommons.org/licenses/by-nc-nd/3.0/
Calcul efficace de la structure des protéines à partir de contacts évolutifs Fabrice Allain © 30 Novembre 2017
Thèse de doctorat en Sciences de la Vie
Rapporteurs : Dr. Pierre Tufféry et Dr. Jean-Christophe Gelly Examinatrice : Dr. Sophie Zinn-Justin
Directeur de thèse : Pr. Michael Nilges
Co-Directeur de thèse : Dr. Benjamin Bardiaux Université Pierre et Marie Curie
École doctorale Complexité du Vivant
Unité de Bioinformatique Structurale Institut Pasteur
25-28 Rue du Docteur Roux 75724 Paris Cedex 15
Résumé
Malgré les progrès constants des techniques expérimentales en biologie structurale, la différence entre le nombre de structures et de séquences protéiques disponibles dans les bases de données ne cesse de croître depuis l’arrivée des technologies de séquençage à haut débit. Les méthodes de prédiction structurale constituent actuellement une alternative relativement fiable pour donner un premier aperçu du repliement natif d’une protéine. Parmi les outils existant, la forte croissance des informations génomiques a remis à l’ordre du jour des techniques modélisant les données capturées au cours de l’évolution. La conservation d’une fonction protéique impose de fortes contraintes sur les interactions impliquées dans le repliement et la fonction. Leur trajectoire évolutive au sein d’une famille protéique se reflète ainsi sous la forme d’un réseau de mutations compensatoires. Une fois détectées, ces dernières peuvent aider à modéliser la conformation d’une protéine. De récents développements ont prouvés l’efficacité à reproduire des cartes de contacts natives entre résidus en partant des corrélations observées dans un alignement de séquences homologues. Cette information a également permis de modéliser des conformations similaires aux structures obtenues expérimentalement.
Les méthodes résolvant la structure tridimensionnelle des protéines à partir des données évolutives présentent encore des limitations dont la détection des contacts faux positifs ou la distinction entre contacts au sein d’une sous unité ou entre deux sous unités. Ces problèmes sont relativement similaires à ceux rencontrés pour la détermination de structure par spectrométrie de Résonnance Magnétique Nucléaire (RMN). Ces expérimentations permettent par exemple de détecter des couples de noyaux proches dans l’espace qui seront ensuite utilisés sous la forme de contraintes de distances pour retrouver une conformation tridimensionnelle de la protéine étudiée.
Le traitement et l’intégration des données de RMN est aujourd’hui un processus clai-rement établi et en grande partie automatisé. Des logiciels comme ARIA (Ambiguous Restraints for Iterative Assignment) et ISD (Inferential Structure Determination) intègrent la détection des faux positifs et l’attribution des contacts intra/inter mono-mériques. ARIA utilise en particulier le concept de contraintes de distances ambiguës et suit un processus itératif afin d’attribuer et d’affiner la liste des noyaux proches dans l’espace pour calculer un ensemble de modèles structuraux en accord avec les données. Ce travail a pour objectif d’adapter et valider l’intérêt de l’approche ARIA pour prédire de novo la structure d’une protéine en utilisant l’information évolutive.
Abstract
Despite tremendous progress in experimental determination of protein three dimen-sional structures, the gap in the international database between protein sequences and structures available in databases is widening rather than diminishing since the explosive growth of sequence information from next generation sequencing technology. As an alternative or complementary solution, computational prediction of protein structures may be able to alleviate the problem by giving preliminary insights of the native fold of a protein.
Among existing tools, the increasing availability of genomic information provides the possibility to capture relevant évolutionary information. Conservation of a specific function implies strong restraints on interacting residues involved in the folding and function. Their evolutionary trajectory across the protein family may be viewed as a network of compensatory mutations. Detecting those co-evolving pairs can then be used to predict the protein conformation. Recent advances has shown the feasibility to predict native contact maps from co-evolving residues observed in multiple sequence alignments. More recently, approaches combining evolutionary contacts with structure calculation methods has also been able to generate protein structures with various level of success.
Some important aspects of these techniques are still to be improved, including the detection of false positive among the predicted contacts or distinction between intra-subunit and inter-monomeric contacts for multi-intra-subunit systems. Here the similarity between the types of information provided by evolutionary contacts and by Nuclear Magnetic Resonance (NMR) spectroscopy is striking. Those experimentations also detects pairs of atoms close in space in order to determine 3D structure of a protein and presents the same difficulties.
In the frame of NMR, efficient and robust approaches are available to analyze, assign and apply distance restraints such as ARIA (Ambiguous Restraints for Iterative Assignment) or ISD (Inferential Structure Determination) from unassigned NMR data. More particularly, ARIA use concepts like Ambiguous Distance Restraints and an iterative protocol to assign NMR data and compute a set of structural models in agreement with those data. This work aims at validating and extending the concept to de novo structure prediction from evolutionary data.
Remerciements
En premier lieu, je souhaite adresser mes remerciements au Pr Michael Nilges pour m’avoir offert le privilège de travailler au sein du laboratoire de Bioinformatique Structurale et d’assurer la direction de cette thèse. Je tiens également à exprimer ma profonde gratitude et ma sincère reconnaissance au Dr Benjamin Bardiaux, co-directeur et initiateur de ce projet. Au-delà de m’accorder sa confiance, il a été tout au long de ces trois années un guide bienveillant et m’a aidé à trouver les solutions adaptées pour avancer.
Je remercie chaleureusement le Dr Pierre Tufféry et le Dr Jean-Christophe Gelly pour avoir bien voulu examiner ce manuscrit de thèse ainsi que le Pr Martin Weigt et le Dr. Sophie Zinn-Justin d’avoir accepté de faire partie de ce jury.
J’exprime aussi mes remerciements à tous les collègues présent et passé de l’unité qui au-delà d’un environnement scientifique enrichissant contribuent à cette ambiance de travail particulièrement sereine. Je voudrais spécialement remercier Nathalie Duclert Savatier, le Dr Tru Hyun, le Dr. Silke Weninger et la secrétaire Maya Um pour votre soutien et nos discussions.
Je profite de ces quelques lignes pour remercier également mon frère et ma mère ainsi que mes proches qui de près ou de loin ont toujours su être à l’écoute dans les moments difficiles. Merci Thibault pour m’avoir offert ton aide, tes encouragements et surtout ton amitié. Je voudrais également remercier en particulier Amel, Luc, Antoine et Marine pour votre soutien sans faille.
Table des matières
Table des figures ix
Liste des tableaux xii
I Introduction 1
1 Modélisation in silico à l’échelle atomique d’une biomolécule 2
1.1 Étude structurale d’une macromolécule biologique . . . 2
1.1.1 Approches expérimentales . . . 2
1.1.2 Modélisation in silico . . . . 8
1.1.3 Limites des approches expérimentales . . . 9
1.2 Prédiction de structure protéique . . . 10
1.2.1 Prédiction comparative . . . 11
1.2.2 Prédiction Ab initio . . . . 12
2 Information co-évolutive en biologie structurale 13 2.1 Classification des familles protéiques . . . 13
2.2 Couplages évolutifs d’un ensemble de séquences homologues . . . . 14
2.2.1 Approches statistiques . . . 15
2.2.2 Apprentissage supervisé (Machine Learning) . . . . 17
2.2.3 Cartes de contact hybrides . . . 18
2.3 Domaines d’application des couplages évolutifs . . . 18
3 Modélisation d’une structure par Résonance Magnétique Nucléaire 19 3.1 Données structurales en RMN . . . 19
3.2 Détermination d’une structure à partir de contraintes expérimentales RMN . . . 20
3.3 ARIA : une approche itérative d’attribution de données RMN et calcul de structure . . . 20
3.3.1 Traitement itératif des données . . . 20
3.3.2 Calcul des structures . . . 23
3.3.3 Validation des structures résolues par RMN . . . 24
4 Objectifs de la thèse 26 4.1 Traitement de l’information de contact . . . 27 4.2 Adaptation du protocole d’ARIA à la prédiction structurale de novo . 27
II Repliement tridimensionnel d’une protéine à partir d’une carte de contact 29
1 Introduction 30
2 Méthodes 31
2.1 Analyse des cartes de contacts . . . 31
2.1.1 Définitions de contact . . . 31
2.1.2 Critères de sélection et d’exclusion empiriques des contacts évolutifs . . . 31
2.1.3 Métriques pour la comparaison des cartes de contacts . . . 33
2.2 Interprétation des cartes de contacts . . . 35
2.2.1 Contraintes de distance évolutives . . . 35
2.2.2 Contraintes de structure secondaire . . . 36
2.3 Schéma de calcul dans ARIA . . . 36
2.3.1 Dynamique moléculaire sous contraintes en recuit simulé . . 37
2.3.2 Analyse des structures . . . 37
2.4 Contexte expérimental . . . 38
2.4.1 Jeu de données EVFold . . . 38
2.4.2 Cartes de contacts de référence . . . 39
3 Résultats 42 3.1 Analyse des cartes de contacts . . . 42
3.1.1 Approche comparative des modèles évolutifs avec les cartes de référence . . . 42
3.1.2 Évaluation des critères de sélection et d’exclusion des contacts évolutifs . . . 44
3.2 Repliement structural à l’aide du protocole ARIA . . . 49
3.2.1 Paramétrisation du protocole de recuit simulé . . . 49
3.2.2 Traitement des contraintes de distances . . . 54
3.2.3 Conformation alternatives récurrentes . . . 57
4 Conclusion 60 III Adaptation d’ARIA à la prédiction de novo 63 1 Introduction 64 2 Matériel et méthodes 66 2.1 Potentiels statistiques . . . 66
2.2 Intégration d’algorithmes de partitionnements dans le procédé itératif 67 2.3 Fonction de score étendue dans ARIA . . . 69
3 Résultats 70
3.1 Extension de la fonction énergétique par des potentiels statistiques . 70
3.2 Analyse en composantes principales des conformations . . . 74
3.3 Application d’algorithmes de partitionnement au processus itératif d’ARIA . . . 75
3.3.1 Partitionnement par les K-moyennes . . . 75
3.3.2 Classification des groupes de structures . . . 80
3.3.3 Attribution automatique des algorithmes de partitionnements 82 3.4 Analyse des conformations . . . 83
4 Conclusions 86 IV Recherche de stratégies complémentaires au traitement de l’information évolutive 89 1 Introduction 90 2 Matériel et méthodes 91 2.1 Contacts liés aux motifs dans les feuillets bêta . . . 91
2.2 Modèles de mixture de gaussiennes sur les distributions de distances 91 2.3 Contraintes expérimentales . . . 92
3 Résultats 94 3.1 Traitements alternatifs de l’information évolutive . . . 94
3.1.1 Distance cible statistique . . . 94
3.1.2 Contacts au voisinage des résidus . . . 97
3.1.3 Contacts aux limites du domaine sur le repliement . . . 98
3.2 Sources d’information complémentaires aux prédictions de contacts . 100 3.2.1 Contacts d’un feuillet bêta dérivés des cartes de contacts prédites100 3.2.2 Combinaison contraintes évolutives et données RMN . . . 100
4 Conclusions 103
V Conclusions 105
VI Annexes 111
A Données supplémentaires 112
B Module ARIAEC 115
Table des figures
I.A Apport des approches expérimentales en biologie structurale . . . 3 I.B Familles de prédiction structurale . . . 11
I.C Illustration des 4 grandes classes de repliement SCOP . . . 14 I.D Représentation schématique d’une corrélation transitive entre deux
résidus . . . 16 I.E Approximation mean field pour dériver les matrices de couplage du
modèle statistique globale . . . 17
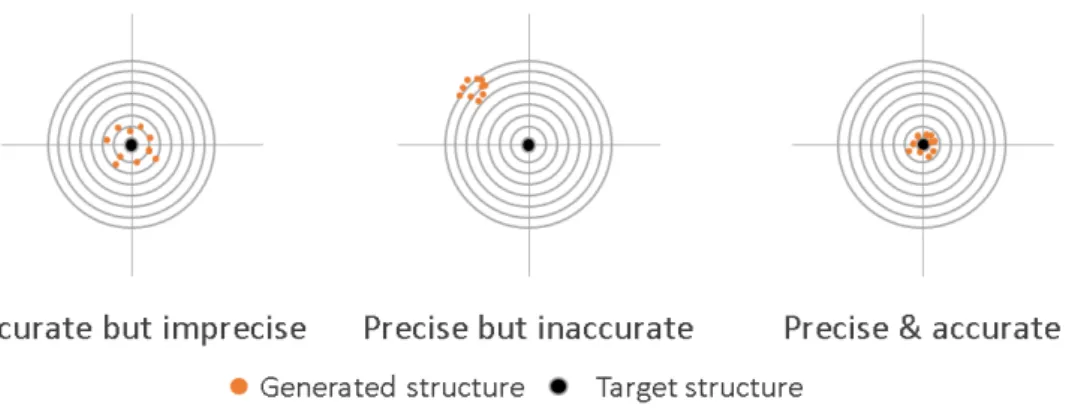
I.F Détermination de structure RMN à l’aide du protocole itératif d’ARIA . 21 I.G Formulation des contraintes de distances simples ou ambiguës . . . 22 I.H Représentation schématique des mesures de précision et justesse . . . 25
I.I Erreurs de prédiction de contact . . . 26
II.A Intégration des cartes de contacts dans le protocole d’ARIA . . . 32
II.B Estimation de la performance des données de prédiction brutes en fonction des cartes natives de contacts éloignés . . . 43 II.C Impact des filtres empiriques sur la qualité des prédictions de contacts 45 II.D Impact des filtres empiriques sur la précision des prédictions de contacts 46 II.E Performance des filtres empiriques en fonction des méthodes de prédiction 47 II.F Précision des filtres empiriques en fonction des méthodes de prédiction 48 II.G Évolution de la précision des conformations en fonction des classes de
repliement . . . 50 II.H Convergence des conformations "natives" et "évolutives" après
modifi-cation du recuit simulé . . . 52 II.I Impact du poids d’EVFold sur la convergence des structures . . . 53 II.J Prédictions à partir de contraintes de distance ambiguës . . . 55 II.K Justesse des ensembles ARIA en fonction du nombre de contraintes
natives sélectionnées à la première itération. . . 58 II.L Images miroires topologiques . . . 59
III.A Potentiel de forces moyennes associé aux liaisons hydrogènes . . . 67
III.B Statistiques de Ramachandran après application du potentiel de forces moyennes . . . 70 III.C Effet du potentiel de Ramachandran sur la convergence des structures 71
III.D Comparaison de la performance des PMF sur la justesse des ensembles sélectionnés par ARIA . . . 73 III.E Analyse en composantes principales des conformations générées par ARIA 74 III.F Répartition des partitions de structures obtenues par K-Means (n=2) . 75 III.G Évolution de la convergence des conformations après partitionnement
par K-Means (n=2) . . . 76 III.H Justesse moyenne des groupes à la dernière itération du protocole ARIA 77 III.I Différence d’énergie par rapport à l’écart de justesse des groupes de
conformations . . . 78 III.J ACP groupes de conformères CD209_HUMAN (contacts natifs) . . . . 79 III.K Relation énergie totale et justesse des structures . . . 80 III.L Classement des groupes de structures par itération avec la fonction de
score standard et étendue d’ARIA . . . 81 III.M Sélection automatique du nombre de groupes par itératio . . . 82 III.N Analyse discriminante linéaire selon les critères qualitatifs et
énergé-tiques des conformations . . . 84 III.O Valeurs moyennes des critères qualitatifs en fonction des classes de
justesse . . . 85
IV.A Modèles de mixture de gaussiennes sur les distributions de distance inter atomique . . . 92
IV.B Distribution des distances entre atomes des structures à haute résolution appartenant à la base de donnée PISCES. . . 94 IV.C Distributions distances cibles fixées empiriquement ou statistiquement 95 IV.D Effet des distances cibles statistiques sur la convergence . . . 96 IV.E Répartition de l’information des groupes de conformation native
incor-rectement sélectionnées . . . 97 IV.F Effet des contraintes au voisinage sur la convergence pour les contraintes
évolutives EVFold . . . 98 IV.G Couverture du domaine CD209_HUMAN . . . 99 IV.H Application des contraintes bbcontacts . . . . 101 IV.I Performance de la méthode en combinant les contacts prédits avec des
données expérimentales . . . 102
IV.J Critères d’information des mixtures de gaussiennes . . . 103
V.A Performance d’ARIA par rapport aux autres approches de prédiction de
novo . . . . 107
VI.A Estimation de la performance des données de prédiction brutes en fonction des cartes natives non filtrées . . . 112 VI.B Poids des critères qualitatifs par rapport aux dimensions de l’analyse
discriminante linéaire . . . 113
VI.C Analyse scores de silhouette groupes après partitionnement par K-moyennes CD209_HUMAN . . . 114
Liste des tableaux
II.A Critères de validité d’un contact . . . 34
II.B Paramètres du protocole de recuit simulé dans ARIA . . . 37
II.C Jeu de données EVFold . . . 40
IV.A Jeu de données EC-NMR . . . 93
IV.B Comparaison avec l’approche EC-NMR . . . 102
I
Introduction
„
In the drama of life on a molecular scale, proteins are where the action is—Arthur M Lesk
Introduction to Protein Architecture [1]
L’étude structurale des bio-molécules joue un rôle important dans la compréhension des mécanismes biologiques. Comprendre leur fonction-nement à l’échelle atomique est aujourd’hui un défi majeur dans le
monde scientifique [2]. Déchiffrer les rôles et fonctions des biomolécules
nécessite l’analyse des structures et des différentes interactions pouvant avoir lieu dans la cellule (protéine-protéine, protéine-acide nucléique, protéine-ligand). Modéliser in silico ces interactions joue également un rôle non négligeable pour l’ingénierie rationnelle de nouveaux médica-ments. Cette partie a pour vocation de donner un aperçu des principales approches utilisées pour élaborer un modèle tridimensionnel d’une pro-téine. Habituellement, cela revient à recourir à des méthodes utilisant des données expérimentales. En parallèle à ces techniques, on peut éga-lement faire appel à des algorithmes de prédiction de structures pour palier aux problèmes rencontrés expérimentalement.
1
Modélisation in silico à l’échelle atomique
d’une biomolécule
1.1
Étude structurale d’une macromolécule
biologique
Les avancées dans le domaine de la biologie structurale sont étroitement liées avec le développement des technologies de visualisation permettant d’explorer les phénomènes invisibles à l’œil nu. Des premiers microscopes optiques au XVIIème siècle à nos jours, la résolution spatiale1des microscopes reposant sur la diffraction de la lumière visible n’a jamais cessée de progresser jusqu’à atteindre quasiment la limite physique théorique (limite de diffraction d’Ernst Abbe2: 0.2 µm) [3-5].
De part sa nature ondulatoire, la lumière comme tout autre radiation nécessite en effet d’avoir une longueur d’onde inférieure aux dimensions de l’objet. L’ordre de grandeur entre les atomes étant de 0 à 5 Å, il est nécessaire d’utiliser des rayonnements électromagnétiques avec des longueurs d’ondes inférieures à celle de la lumière visible (200 nm) pour visualiser les structures au niveau atomique. Les techniques expérimentales utilisées de nos jours reposent ainsi sur d’autres types de radiations adaptées à l’échelle moléculaire comme les photons provenant de rayons X en cristallographie ou encore des faisceaux d’électrons pour la microscopie électronique. En parallèle des procédés d’imagerie cellulaire, la spectroscopie par Résonance Magnétique Nucléaire joue aussi un rôle privilégié pour analyser et caractériser la structure et la dynamique d’un système.
1.1.1
Approches expérimentales
Les approches expérimentales ont suivit une rapide évolution au cours du siècle dernier aussi bien en efficacité qu’en fiabilité à déterminer les coordonnées ato-miques d’un système. Les avancées techniques ont ainsi permis l’automatisation progressive dans les étapes de préparation des échantillons (extraction, production, purification, . . .), les protocoles d’acquisition et l’analyse des données. L’étroite
rela-1. Distance à laquelle deux points peuvent être perçus par un photorécepteur
2. Théorie sur la distance maximale permettant de distinguer deux points à l’aide d’un microscope photonique. Cette limite théorique imposée par le phénomène de diffraction est proportionnelle à la longueur d’onde du rayonnement utilisé. En pratique, elle correspond avec les système optiques modernes à environ la moitié de la longueur d’onde soit 200 nm pour la lumière visible
1973 1975 1977 1979 1981 1983 1985 1987 1989 1991 1993 1995 1997 1999 2001 2003 2005 2007 2009 2011 2013 2015 Année 100 101 102 103 104
Structures protéiques publiées par an (PDB)
Rayons X RMN EMTotal
aÉvolution de l’apport par an des approches expérimentales
1973 1975 1977 1979 1981 1983 1985 1987 1989 1991 1993 1995 1997 1999 2001 2003 2005 2007 2009 2011 2013 2015 0 20000 40000 60000 80000 100000 120000
Structures protéiques publiées (PDB)
b Nombre de structures publiées
Figure I.A:Apport des approches expérimentales en biologie structurale. La part de la
cristallographie reste la plus importante avec dix fois plus de structures déposées
par rapport aux deux autres approches (I.Aa). Le nombre de structures déposées
au total dans la PDB croît de manière exponentielle depuis la fin des années 1990 (I.Ab).
tion entretenue entre la structure et la fonction d’une protéine a été à l’origine de projets en génomique structural et protéomique d’envergure internationale (Struc-tural Genomics Consortium [6], Protein Structure Initiative [7], Human Proteome Organization [8]) au début des années 2000 qui ont également fortement contribué au développement dans le domaine. Le nombre de structures protéiques résolues a ainsi connu une forte augmentation ces deux dernière décennies passant d’environ 1000 structures publiées par an à plus de 10 000 en 2016 (Fig.I.A).
Cristallographie aux rayons X
Historiquement, les premières structures de bio-molécules résolues proviennent de l’interprétation de spectres de diffraction produits par des cristaux de protéines irradiées aux rayons X. L’idée d’utiliser ce type de rayonnement pour déterminer l’organisation tridimensionnelle d’un cristal fut suggérée la première fois par Max von Lau en 1912 [9]. William Henry Bragg et son fils Lawrence Bragg [10] validèrent cette hypothèse en 1913 en reconstituant la structure du chlorure de sodium à partir
Chapitre 1 Modélisation in silico à l’échelle atomique d’une
de son spectre de diffraction. Cependant, l’utilisation de la cristallographie aux rayons X ne fut pas tout de suite envisagée comme une solution viable pour les protéines. Ces dernières étant considérées comme des colloïdes [11] de structure aléatoires. Cette perspective a commencée à changer lorsque Sumner en 1928 obtenu pour la première fois le cristal de l’uréase [12], une enzyme catalysant l’hydrolyse de l’urée en dioxyde de carbone et ammoniac. En 1934, Bernal et Hodgkin réussirent à obtenir un spectre de diffraction à partir de la pepsine [13]. Il fallut attendre plusieurs années pour résoudre les différents problèmes techniques et théoriques avant de voir la première structure de protéine résolue par cristallographie aux rayons X avec les travaux de John Kendrew sur la myoglobine [14] suivit un an plus tard avec la structure de l’hémoglobine par l’équipe de Max Perutz [15, 16]. Dès lors, cette technique est devenue essentielle et n’a jamais cessée de gagner en efficacité comme l’illustre la part importante de structures résolues par cette méthode aujourd’hui (Fig.I.Ab).
Le principe de la radiocristallographie est de reconstituer une carte de densité asso-ciée à un spectre de diffraction des électrons. Un faisceau de rayons X est envoyé sur un échantillon monocristallin provoquant une déformation du nuage électronique au contact des atomes. Cette déformation sera à l’origine d’une nouvelle radiation dans toutes les directions de l’espace dont l’énergie sera similaire au rayonnement incident (diffusion élastique dite de Rayleigh dans cette situation). La périodicité du réseau cristallin permet aux ondes diffractées par le réseau d’atomes de se com-biner entre elles et d’amplifier le signal si elles ont la même phase (interférence constructive) soit en opposition de phase et réduire le signal (interférence destruc-tive). L’intensité des photons capturée par le spectre de diffraction correspond ainsi à la somme des interférences constructives. L’amplitude d’une onde diffusée étant proportionnelle au nombre d’électrons traversés, elles sont donc caractéristiques de la nature des atomes traversés par les rayons X. Pour reconstituer la structure de la molécule, il est cependant nécessaire de déterminer la phase des ondes projetées sur le spectre de diffraction. Cette information n’est malheureusement pas disponible ex-périmentalement et retrouver les différentes phases constitue l’étape la plus délicate pour retrouver la structure associée au cliché. Il est toutefois possible de reconsti-tuer l’information via des méthodes statistiques ou des approches nécessitant une connaissance de la composition chimique du système étudié au préalable (méthode de l’atome lourd, remplacement moléculaire, diffusion anomale, . . .).
La cristallographie aux rayons X n’étant pas limitée par la taille de la molécule, elle est souvent choisit en premier dès lors qu’on cherche à résoudre la structure. La nécessité d’un cristal en quantité suffisante et présentant un pouvoir diffractant élevé reste une étape difficile. Les obstacles rencontrées pour aboutir à des cristaux peuvent être de différentes nature et requiert un large éventail d’expertises. Quelques exemples sont listés ci dessous :
•
Production de l’échantillon : la synthèse peptidique, l’extraction et la purifi-cation de la protéine peut présenter des complipurifi-cations notamment lorsqu’il y a un manque d’expression ou de solubilité de la protéine cible.•
Cristallogénèse : cette étape est souvent la plus difficile à mettre en place. L’expérimentation est en effet particulièrement sensible à la taille et la pureté des cristaux. Les rayons X détruisant le cristal, il est également important d’en avoir une quantité suffisante pour observer un signal. La difficulté ici est de définir les conditions optimales de cristallisation. Ces dernières variant d’une protéine à l’autre, la recherche bien qu’en grande partie automatisée ces dernières années est encore principalement empirique. Selon les conditions, on peut retrouver des situation d’enchevêtrement de cristaux (formation de macles) ou la formation de pseudo symétries dans le cristal. Plusieurs centaines voir des milliers de conditions sont évaluées en général.•
Collecte des données : le problème ici concerne essentiellement l’attribution des phases associées aux spectres de diffraction. Parmi les solutions existante, une pratique courante passe par des méthodes de diffusion anomales à l’aide de sources de rayonnements synchrotron à longueur d’onde multiples.Résonance Magnétique Nucléaire
La spectroscopie par Résonance Magnétique Nucléaire (RMN) est une technique apportant une information sur la structure et la dynamique à l’échelle atomique des molécules aussi bien en phase liquide que solide. La découverte de la résonance magnétique nucléaire date de 1946 par deux groupes indépendants [17, 18] reposant entre autre sur les observations faites par l’équipe de Rabi des propriétés magnétiques de certains noyaux atomiques en 1938 [19]. Parmi les principales étapes qui ont valu l’introduction et le développement de cette méthode dans le domaine de la chimie structurale, on peut citer la découverte du déplacement chimique en 1950 [20, 21], la description théorique du transfert d’aimantation par couplage dipolaire entre deux noyaux (effet Overhauser) en 1953 [22] ou encore la publication du premier spectre de protéine en 1957 [23]. L’effet Overhauser étant étroitement relié à la distance entre deux noyaux, les efforts se sont concentrés par la suite sur le problème d’attribution des signaux à un groupement chimique. L’introduction de spectres combinant plusieurs dimensions[24] est une autre grande avancée dans le domaine permettant de détecter différentes interactions avec une résolution accrue. Wüthrich et ses collègues furent les premiers à mettre en place un protocole combinant les informations provenant de la RMN pour déterminer la structure d’une protéine en phase liquide [25].
Chapitre 1 Modélisation in silico à l’échelle atomique d’une
La spectroscopie RMN s’appuie sur l’étude de l’absorption et la rediffusion de ra-diations électromagnétiques d’un nombre limité d’isotopes (1H,2H,13C,14N,15N,
. . .) soumis à un champ magnétique externe. L’isotope le plus utilisé en RMN reste le noyau d’hydrogène (1H) contenant un unique proton. La possibilité d’étudier un isotope par résonance magnétique est directement liée à la notion de spin nucléaire. Le spin nucléaire est une propriété quantique intrinsèque des particules élémentaires au même titre que la masse ou la charge. Lorsque le spin d’un noyau est non nul, le mouvement circulaire des charges des particules élémentaires dans le noyau génère un champ magnétique représenté par une grandeur vectorielle nommée moment magnétique nucléaire. La particularité du moment magnétique nucléaire est qu’il n’existe que dans deux directions possibles (états de spin α et β).
En dehors de toute perturbation magnétique, l’énergie associée aux dipôles magné-tiques est identique et leur orientation répartie de manière aléatoire dans l’espace. La première partie d’une expérimentation en RMN est d’appliquer un champ magné-tique externe uniforme. En s’alignant avec ce champ, les axes de rotation des noyaux vont précesser autour du champ magnétique dans deux orientations différentes divisant les noyaux en deux populations (effet Zeeman). L’orientation parallèle (état α) correspond à l’état énergétique le plus faible. La seconde population étant l’orien-tation antiparallèle (état β) du moment magnétique avec le champ. La fréquence de précession (fréquence de Larmor) autour de l’axe du champ est directement reliée à l’énergie de transition entre les deux états. L’énergie associée aux deux états étant elle même proportionnelle au rapport gyromagnétique et donc à la nature du noyau et de son nuage électronique.
La spectroscopie de résonance magnétique nucléaire s’intéresse ainsi au phénomène d’absorption d’une radiation électromagnétique dont la fréquence (l’énergie d’un photon étant proportionnelle à sa fréquence) correspond à la différence d’énergie entre les deux états. Les spins nucléaires entrent donc en résonance avec une radiation électromagnétique spécifique en fonction de leur fréquence de précession. Les spectres sont habituellement enregistrés en modifiant soit l’intensité du champ de force soit la fréquence de la radiation électromagnétique pour pouvoir observer l’ensemble des isotopes de la molécule.
Les expérimentations partant du principe de résonance permettent d’obtenir un ensemble de caractéristiques structurales sur la molécule étudiée. Ces dernières serviront de contraintes pour calculer un ensemble de conformères par l’intermé-diaire d’algorithmes de géométrie de distance ou par dynamique moléculaire dans un espace réel. L’optimisation des structures suit habituellement un protocole de recuit simulé. L’étape d’attribution des mesures d’effets Effet Overhauser Nucléaire permettant de relier tous les pics croisés à des couples de noyaux dans un spectre est l’étape qui nécessite le plus de temps, d’expertise (la complexité du spectre étant
directement liée à la taille de la protéine) et est la plus importante pour la détermi-nation de structure. Il n’est en général pas possible de pouvoir attribuer l’ensemble des pics croisés sans aucune ambiguïté à cause de la précision expérimentale limitée sur la position des pics et les valeurs de déplacements chimiques augmentant ainsi le nombre de possibilités d’attributions pour un pic donné. Le recouvrement de certains pics croisés sur les spectres, la possibilité d’observer des artefacts ou encore l’absence de signal à cause d’une relaxation trop rapide au cours de l’expérimentation sont éga-lement des facteurs qui compliquent cette tâche. Ces différents problèmes expliquent l’utilisation de logiciels permettant d’attribuer de façon automatique des spectres et de calculer les structures tri-dimensionnelles, comme CANDID/CYANA[26, 27], AUTOSTRUCTURE[28] ou ARIA [29-34].
Le principal avantage par rapport aux autres approches expérimentales est de pouvoir étudier les structures en solution ou en phase solide ce qui correspond donc à un milieu plus proche de l’état physiologique par rapport à la cristallographie aux rayons X. De plus, l’énergie des photons résonant avec les états de spins soumis à un champ magnétique de forte intensité étant relativement faible. Les radiations utilisées par la spectroscopie en RMN sont peu énergétiques (avec des longueurs d’ondes d’environ 1m). Contrairement à la diffractométrie aux rayons X, cette méthode est peu destructrice.
Déterminer une structure en RMN reste cependant une tâche longue et complexe. Le rapport signal sur bruit et la complexité des spectres augmentant avec la taille de la molécule étudiée, les expérimentations en RMN sont essentiellement concentrées sur des complexes dont le poids n’excède que très rarement les 25 kDa. L’apport d’in-formations complémentaires comme les mesures de couplages dipolaires résiduels sur des molécules en partie orientées permet de relever cette limite aux alentours de 50 kDa [35]. De plus, la mise en place d’une expérience de spectroscopie RMN nécessite l’utilisation de solutions fortement concentrées (de l’ordre du millimolaire pour des protéines d’environ 15 kDa).
Cryo microscopie électronique
Développé par l’équipe de Knoll dans les années 1930 [36], la microscopie électro-nique concentre des faisceaux d’électrons pour interagir avec la matière à la manière d’un microscope photonique classique. Les électrons propulsés à grande vitesse sur l’échantillon possèdent une longueur d’onde de l’ordre du picomètre permettant l’observation de la matière à l’échelle atomique. Cette technique fut la première à fournir des images à la résolution moléculaire comme l’observation de particules de virus par coloration négative en 1959 [37]. Les méthodes de traitement et de reconstruction des images tridimensionnelles ont par la suite permit de détermi-ner la structure d’une partie du bactériophage T4 en 1968 [38]. Les imperfections
Chapitre 1 Modélisation in silico à l’échelle atomique d’une
causées notamment par l’échantillon et les instruments ne permit pas d’atteindre de résolution dépassant les 7 Å [39] avant le début des années 90 avec un modèle atteignant 3.5 Å de résolution [40] (bien que limitée à certains plans du complexe). Un développement plus récent dans le domaine concerne l’analyse en particule unique où les molécules sont bloquées dans une orientation aléatoire à l’intérieur d’une glace vitreuse [41]. Dernièrement, une carte d’une enzyme bactérienne, la beta galactosidase a atteint une résolution de 2.2 Å [42].
La qualité de préparation des échantillons et l’intensité du faisceau d’électrons sont des facteurs clés pour atteindre une résolution atomique. Ces derniers doivent être suffisamment fin pour pouvoir observer une interférence et le faisceau suffisamment intense pour diminuer le ratio bruit/signal dans les images obtenues. Le principal risque en augmentant la puissance du faisceau est d’endommager l’échantillon. Pour palier à ce problème, on diminue la température de l’échantillon (cryo-microscopie) dans le but de protéger et maintenir les échantillons dans un état stable.
Techniques complémentaires
Caractériser des complexes moléculaire de taille importante ou possédant une forte flexibilité est souvent reliée à une perte en résolution et qualité des données. En com-pléments des approches classiques à haute résolution, d’autres méthodes permettent d’obtenir des informations plus ou moins précises sur la forme ou les interactions d’un complexe. Un des exemples les plus représentatifs est la diffusion des rayons X ou neutrons aux petits angles. Ces deux méthodes sont couramment utilisée de nos jours pour obtenir rapidement des informations à plus basse résolution. Elles ne permettent cependant pas d’atteindre une résolution atomique (la résolution maximale étant autour de 13 Å) mais donnent essentiellement une information globale sur la forme de la structure.
1.1.2
Modélisation in silico
Optimiser les coordonnées spatiales à partir des contraintes géométriques obtenues expérimentalement nécessite une représentation réaliste du système tenant compte des propriétés à la fois physiques et chimiques du milieu expérimental mais aussi de l’incertitude présente dans les données. Ces dernières pouvant par exemple provenir d’un manque de précision des mesures ou de données structurales hétérogènes.
Simuler le comportement de ces systèmes ressort ici du domaine de la mécanique moléculaire qui fait abstraction des mouvements des nuages électrostatiques pour décrire les différents états d’un système. Deux types d’approches existent pour cela, l’approche probabiliste avec les méthodes d’échantillonnage de Monte Carlo et
celle déterministe regroupant les simulations de dynamiques moléculaire. Dans ces algorithmes, les états successifs du système reposent sur les lois du mouvement de Newton. L’intégration de la seconde loi permet en particulier de décrire la trajectoire des particules par le rapport entre les forces appliquées au système sur leur masse.
Au vu de la complexité de la surface d’énergie associée aux systèmes biologiques, les avancées technologiques en informatique ont joué un rôle déterminant dans le développement des méthodes de simulation. La première formulation de simulation moléculaire par l’algorithme de recherche Monte Carlo date de 1953 [43]. La preuve du concept apparut plus tardivement avec la simulation de l’eau au début des années 70 [44] suivit à la fin de la même décennie de la première modélisation d’une protéine : l’inhibiteur de la trypsine pancréatique bovine (BPTI) [45].
L’apport de la mécanique moléculaire joue actuellement un rôle particulièrement important pour comprendre la structure des complexes macromoléculaires, étudier leur dynamique ou encore définir les interactions avec l’environnement cellulaire. Ces connaissances sont particulièrement importante de nos jours pour la concep-tion de nouveau traitements thérapeutiques ou encore comprendre l’appariconcep-tion de mécanismes de résistances.
1.1.3
Limites des approches expérimentales
Deux types de démarche s’opposent actuellement dans la recherche sur les structures de macromolécules biologiques. La première s’inscrit dans des projets de grande envergure en génomique structurale [46, 47] et à plus long terme dans la reconstruc-tion de réseaux métaboliques [48]. Dans ces méthodes, on recherche à résoudre un maximum de structures encodées par un génome à l’aide de protocoles automatisés. Une bonne partie des protocoles de détermination de structure décrit précédemment se prêtent ainsi à une automatisation à grande échelle. Le clonage des gènes, l’ex-pression et purification des échantillon, la collecte des données sont autant d’étapes utilisant des systèmes robotiques.
La deuxième approche plus spécifique concerne la recherche de conformations de protéines dans des conditions précises pour répondre à une question biologique donnée. Contrairement à la génomique structurale, on s’intéresse ici à résoudre une structure précise associée à des contraintes expérimentales spécifiques. De plus en plus de projets en biologie structurale tendent actuellement vers l’intégration de données provenant de plusieurs méthodes expérimentales durant le processus de modélisation. Les approches décrites précédemment ne respectent pas forcément les même conditions expérimentales. Ces dernières pouvant être plus ou moins éloignées du milieu in cellulo et in vivo. De plus elle ne se positionnent pas forcément aux
Chapitre 1 Modélisation in silico à l’échelle atomique d’une
mêmes échelles de résolution ni de temps. Le problème du choix d’une ou plusieurs méthodes pour élucider la structure du système reste particulièrement critique. La proportion relativement stable observée entre les différentes techniques témoigne de cette complémentarité.
Modéliser les protéomes de plusieurs milliers d’espèces est un projet ambitieux mais irréalisable à l’heure actuelle. Le nombre de structure déterminées par année tend a stagner contrairement à la croissance exponentielle du nombre de séquences protéiques depuis l’arrivée des techniques de séquençage à haut débit (voir figure 1.C)[49, 50]. L’obtention d’une structure par les procédés automatisés en génomique structurale reste encore largement aléatoire et ne peut empêcher cet écart de se creuser. De plus, la résolution expérimentale d’une structure peut durer de plusieurs mois à des années en fonction de la protéine étudiée (taille, purification, conditions d’extractions, cristallisation, . . .). Malgré un investissement important dans le do-maine, ces difficultés propres à chaque méthode sont toujours présentes de nos jours et peuvent mener au ralentissement voir à l’échec à déterminer une structure.
Derrière cette quantité d’information phénoménale apportée par la génomique, pré-dire une structure à partir de l’information contenue dans les séquences protéiques apparaît nécessaire pour accélérer la conception de modèles expérimentaux mais également pour combler en partie ce manque d’information. Les méthodes actuelles se distinguent essentiellement par des approches dites comparatives ou ab initio [51, 52].
1.2
Prédiction de structure protéique
Prédire la structure d’une protéine avec pour unique matériau de base l’information contenue dans la séquence peptidique reste encore une des grandes questions à résoudre actuellement [53]. Le bénéfice d’utiliser ces approches prend tout son sens dans le cadre de la génomique structurale mais également lorsque les données expérimentales sont imprécises ou incomplètes. Les méthodes de prédiction d’une structure en l’absence de données expérimentales peuvent actuellement être classées dans deux grandes catégories. Parmi les deux familles de prédiction illustrées dans la figureI.B, on peut proposer un échelon supplémentaire pour distinguer les méthodes comparatives. Cette différence s’établit selon la provenance du modèle structural. Les méthodes ab initio se distinguent quant à elles principalement par le type de contraintes apportées au système. Ces dernières pouvant être basées uniquement sur les connaissances empiriques du système ou bien combinées avec d’autres contraintes géométriques prédites.
Figure I.B:Familles de prédiction structurale. La prédiction de l’arrangement spatial d’une protéine soulève à la fois la question de l’appartenance d’un acide aminé aux classes de structures secondaires (hélice α, brin β, coude) mais également sur les interactions entre acides aminés pouvant caractériser l’organisation tridi-mensionnelle du domaine (structure tertiaire). Les méthodes de modélisation comparative (par homologie ou enfilage) ou ab initio représentent les deux grandes stratégies proposées dans le domaine.
1.2.1
Prédiction comparative
Dans les algorithmes de prédiction ab initio, le nombre de degrés de liberté croît rapidement en fonction de la longueur de séquence. Il est donc nécessaire d’utiliser des méthodes alternatives pour restreindre l’espace de recherche en modélisation. Une solution particulièrement efficace actuellement s’appuie sur la comparaison de modèles structuraux pour prédire un ensemble de conformations.
Recherche par homologie
Considérée actuellement comme une des approches les plus performantes, la mo-délisation comparative par homologie consiste à sélectionner une (ou plusieurs) structures généralement déterminées par des méthodes expérimentales à haute résolution et appartenant à la même famille protéique pour servir de base à la construction d’un modèle. On s’intéresse dès lors aux séquences homologues parta-geant des acides aminés identiques ou similaires à la protéine d’intérêt. Ce degré de conservation constitue le principal facteur limitant dans certains cas où on ne peut trouver de modèles suffisamment proches de la protéine recherchée.
Les différents outils disponibles pour ce type de prédiction se démarquent essen-tiellement par le type de modélisation utilisée. On distingue ainsi des prédictions reposant sur l’assemblage de fragments conservés ou sur l’optimisation par dyna-mique moléculaire restreinte par un ensemble de contraintes spatiales dérivées de l’ensemble des modèles structuraux servant de base pour la conception [54]. Parmi les algorithmes populaires proposant la modélisation comparative par homologie, on retrouve la plateforme SWISS-MODEL [55] ou encore la suite ROSETTA [56].
Chapitre 1 Modélisation in silico à l’échelle atomique d’une
Reconnaissance d’une classe de repliement (enfilage)
Dans le cas où il n’existe pas ou peu d’information structurale homologue sur lequel se projeter pour définir une conformation, la prédiction peut passer par des bases de données structurales plus généralistes. Dans ces situations, la structure est simplement suggérée en évaluant le degré de compatibilité de la séquence d’intérêt avec les modèles ou fragments appartenant à différentes classes de repliements. Les serveurs tel qu’I-TASSER [57] et RaptorX [58] proposent par exemple cette alternative.
1.2.2
Prédiction Ab initio
L’héritage d’Anfinsen [59, 60] suggère qu’une protéine dans son environnement natif se replie in cellulo vers une conformation thermodynamiquement stable (minimum d’énergie libre de Gibbs3) dans une échelle de temps allant de la milliseconde à plusieurs heures. Cette observation est à l’origine même des méthodes de prédiction dites ab initio où on cherche à reproduire la forme repliée d’une protéine uniquement par mécanique moléculaire. La structure prédite étant la forme associée au minimum d’énergie potentielle des forces appliquées au système.
L’avantage des méthodes ab initio réside dans l’absence de connaissance statistique à l’exception des propriétés physiques et chimiques simulées par le champ de force. Cet avantage constitue aussi sa principale faiblesse, une telle démarche dépend entièrement de l’algorithme de recherche utilisé et de la complexité de la surface d’énergie potentielle. Cette dernière augmentant considérablement le temps de calcul dès lors qu’on augmente la taille du complexe étudié.
La principale difficulté est de définir une méthode de recherche capable de refléter les conditions de repliement. Une exploration exhaustive du système ne peut pas être envisagée ni définir une représentation réaliste des conditions de repliement d’une protéine comme l’illustre le paradoxe de Levinthal [61]. La solution n’est pas triviale et possède encore à ce jour de nombreuses zones d’ombres. C’est notamment le cas des protéines dites désordonnées où la recherche des conditions de repliement est toujours d’actualité [62].
3. Cette formulation de l’énergie libre est plus proche des conditions expérimentales conduites à pression et température constante
2
Information co-évolutive en biologie
structurale
Prédire une structure à partir uniquement de la séquence est une des énigmes qui préoccupe le domaine de la biologie structurale depuis plus d’un demi-siècle. Tran-cher ce nœud gordien revient à modéliser exhaustivement les conformations d’un système à la manière des méthodes ab initio. Dans ces dernières, l’exploration reste cependant stochastique et demande de vastes ressources informatiques rendant l’ap-proche envisageable que pour des protéines de très petite taille. Pour restreindre cet espace de recherche, l’utilisation de paramètres structuraux apparaît nécessaire.
La séquence primaire en acide aminé d’une protéine permet à elle seule d’apporter plusieurs des propriétés structurales comme l’accessibilité au solvant ou encore les structures secondaires. En combinant cette information avec les motifs de corrélation dans les séquence homologues, il est possible d’étendre la prédiction a un plus grand nombre de caractéristiques dont l’exemple le plus représentatif est la construction d’une carte de contact entre résidus.
2.1
Classification des familles protéiques
Les régions associées à une fonction protéique sont couramment désignées sous l’appellation de domaines protéiques et partagent la particularité de pouvoir se re-plier de manière autonome [42, 63]. Ces régions considérablement plus conservées par rapport aux séquences primaires d’une protéine [64] constituent généralement l’échelon de base dans les classifications structurales. L’approche hiérarchique SCOP [65] regroupe ainsi les domaines qui partagent une fonction commune en familles et super familles selon le degré d’identité des séquences. Les échelons suivant cor-respondent respectivement à l’arrangement topologique des éléments de structures secondaires du domaine et la catégorie de repliement subdivisée en cinq catégories : α, α+β,α/β, βou domaine multiple (voir Fig.I.C).
Cette classification peut être partiellement manuelle dans le cas de l’approche hiérarchique SCOP ou bien complètement automatisée avec la classification CATH [66]. Une approche hiérarchique paraissant peu réaliste pour représenter l’ensemble du protéome, des alternatives sont actuellement explorées pour catégoriser les éléments structuraux des protéines comme le montre les versions plus récentes de la classification SCOP [67, 68].
a b
c
d
Figure I.C: Illustration des 4 grandes classes de repliement dans la classification SCOP :
α(I.Ca), β (I.Cb), α/β (I.Cc), α+β (I.Cd),
2.2
Couplages évolutifs d’un ensemble de
séquences homologues
L’histoire évolutive au sein d’une même famille de protéine impose de fortes contraintes au niveau du ou des domaines qui la compose. Zuckerkandl et Pau-ling [69] ont été parmi les premiers à observer une accumulation de divergences essentiellement dans les sites fonctionnels entre les différentes familles protéiques. L’étroite relation entre la fonction et la structure d’une protéine [70] implique de fortes contraintes structurales observables par le réseau de mutations compensatoires au cours de l’évolution d’une protéine [71-75]. En d’autre termes, les résidus impli-qués dans des interactions énergiquement favorables et nécessaires à la conservation d’une fonction auront tendance à co-évoluer ensemble.
Une application directe de ce concept est la prédiction de cartes de contacts à partir des corrélations observées entre résidus dans un alignement de séquences homo-logues. Dès lors qu’il existe suffisamment d’information dans la famille, ces réseaux d’interactions prédits permettent alors de retrouver une structure indépendamment de toute donnée structurale au même titre que les méthodes de prédiction ab initio.
Le nombre de séquences dans les familles protéiques joue donc un rôle essentiel dans la prédiction et explique en grande partie l’absence de méthodes suffisamment robustes et précises avant l’arrivée des techniques de séquençage à haut débit. Ce nombre de séquence nécessaires étant fixé dans les premières méthodes autour de cinq fois la longueur de la séquence cible [76]. Cette condition reste toutefois sujette à discussion et varie en fonction des algorithmes proposés. La métrique Meff mesurant le nombre de séquences homologues non redondantes étant actuellement une des plus populaires pour estimer la qualité d’information de séquence en vue d’une prédiction de contacts [77].
2.2.1
Approches statistiques
Modèle local
D’un point de vue historique, les premières méthodes de prédiction sont des modèles statistiques basés sur les fréquences observées dans les séquences d’une famille protéique pour dériver un ensemble de contraintes spatiales [78]. Dans ces modèles surnommés locaux, les corrélations observées dans l’alignement de séquence sont considérées indépendantes les unes des autres. Cette approximation permet de formaliser des modèles relativement simples comme la mesure Statistical Coupling
Analysis (SCA) [79] ou l’information mutuelle M Iij [80]. Cette dernière dérivée
de l’entropie de Boltzmann-Shannon [81] évalue ainsi l’écart fij(Ai,k, Aj,k) entre
les fréquences observées des acides aminés aux positions i et j pour l’ensemble des états k et l (q = 21 valeurs possibles correspondant aux 20 acides aminés et un état supplémentaire pour inclure les décalage au niveau de l’alignement) par rapport à la distribution théorique fi(Ai,k)fj(Aj,k)lorsque les deux positions sont
indépendantes : Mij = q X k=1 q X l=1 fij(Ai,k, Aj,k)ln fij(Ai,k, Aj,k) fi(Ai,k)fj(Aj,k) ! (2.1)
Bien qu’elles soient efficaces pour prédire en partie une carte de contact de référence [80, 82, 83], ces approches ne traduisent pas nécessairement une interaction directe entre les deux résidus [84]. Un faible taux de mutation ou encore un échantillon-nage non équilibré dans les alignements de séquences peut par exemple biaiser les fréquences observées.
La principale source d’incohérence entre une carte de contact native et les modèles de prédictions locaux à l’origine de faux positifs correspond cependant aux corrélations indirectes où les contacts prédits partagent un acide aminé en commun. L’exemple le plus simple correspond à un triplet de résidus ou deux acides aminés peuvent être prédits en contacts dès lors qu’ils partagent un contact avec un troisième acide aminé dans la protéine native (voir Fig.I.D).
Modèle global
Afin de distinguer les couples de résidus directement liés dans le réseau de corrélation, une solution est de reformuler le modèle sur l’ensemble de la séquence. Les scores de prédictions associés à deux positions deviennent dès lors des paramètres relatifs au modèle statistique de la famille protéique. Cette problématique revient à définir
Figure I.D: Exemple de corrélation transitive entre deux résidus : bien que le résidu glycine (rouge) ne soit pas à proximité du résidu valine (vert), ces derniers peuvent apparaître corrélés dans les modèles co-évolutifs car ils partagent un contact en commun avec le résidu leucine (Bleu).
une densité de probabilité conjointe respectant les corrélations observées dans l’alignement comme contraintes. Définir avec exactitude cette distribution pose cependant un inconvénient majeur en terme de calcul. Le nombre de paramètres à estimer est ici égal à qLoù L est la longueur de la séquence cible et q le nombre
d’états possibles (21 en incluant les ouvertures dans l’alignement). Pour réduire les paramètres, une simplification est de diminuer le nombre de contraintes aux fréquences associées respectivement à une (fi(Ai)) et deux positions (fij(Ai, Aj)).
Pi(Ai) ≡ X {Ak=1...q}|k6=i P (A1, ..., AL) = fi(Ai) Pij(Ai, Aj) ≡ X {Ak=1...q}|k6=i,j P (A1, ..., AL) = fij(Ai, Aj) (2.2)
Le choix des distributions dans ces méthodes repose sur la maximisation de l’entropie de Shannon. Cette solution dérivée du modèle physique Hopfield-Potts prend la
forme d’une fonction de probabilité jointe pour la séquence cible P (A1...AL) en
intégrant des paramètres scalaires pour respecter les conditions de départ :
P (A1...AL) = 1 Zexp X 1≤i<j≤L eij(Ai, Aj) + Σ 1≤i<≤Lhi(Ai) Z = X {Ai|i=1..L} exp X 1≤i<j≤L eij(Ai, Aj) + X 1≤i<≤L hi(Ai) (2.3)
Estimer les matrices associés à une hi(Ai)et deux positions eij(Ai, Aj)pour
retrou-ver les fréquences observées reste également une tâche couteuse en terme de calcul et nécessite d’avoir recours à un degré d’approximation supplémentaire. C’est cette dernière étape qui explique le nombre important d’approches statistiques reposant sur ce même modèle statistique. Les premières méthodes approximant ces para-mètres par les algorithmes de message passing (mpDCA [85]) soit par l’inversion de la matrice de corrélation dans le cas des approches mfDCA [86] et PsiCov [87]. L’al-gorithme mfDCA est présenté en guise d’exemple dans la figureI.E. Plus récemment, les méthodes comme plmDCA [88] et GREMLIN [89] reposent sur la maximisation d’une fonction de vraisemblance.
Figure I.E:Approximation mean field pour dériver les matrices de couplage du modèle
statistique globale. Les matrices de couplages entre deux positions distinctes
iet j dans l’alignement de séquence sont ici approximées par inversion de la
matrice de corrélation. Cette dernière étant construite à partir des fréquences observées pour chaque type d’acide aminé.
2.2.2
Apprentissage supervisé (Machine Learning)
Tirant profit des algorithmes d’analyses de données à grande échelle, ces méthodes reposent sur la classification supervisée de caractéristiques physiques et chimiques propre à la séquence avec les données évolutives de la famille protéique pour construire les modèles de prédictions. Ce domaine est actuellement un des plus prolifiques au vu du nombre conséquent de publications par année. Les principales variations entre ces approches concernent les paramètres utilisés et l’algorithme
utilisé d’apprentissage. Dans la liste on retrouve des applications de la méthode d’apprentissage SVM avec SVMcon [90], les réseaux de neurones convolutifs avec CNNcon [91] ou encore la méthodes des arbres aléatoires dans PhyCMAP [77]. Les modèles récents s’orientent vers l’utilisation d’algorithmes d’apprentissages supervisés dits profonds (ou deep learning) possédant des couches d’abstraction supplémentaires par rapport aux algorithmes initiaux [92, 93].
2.2.3
Cartes de contact hybrides
A l’interface entre les deux autres concepts, les algorithmes hybrides combinent les prédictions des méthodes statistiques avec les cartes provenant des modèles d’ap-prentissage supervisé. La qualité d’information contenue dans une famille protéique étant définie par l’algorithme d’alignement de séquence associé, d’autres méthodes vont jusqu’à ajouter les paramètres de l’algorithme d’alignement dans l’étape d’ap-prentissage comme l’approche PconsC [94-96] et plus récemment MetaPsicov [97] pour prendre ainsi compte des différentes possibilités d’alignements. Ces méthodes ont en général un meilleur rapport signal sur bruit mais n’apportent pas nécessai-rement de changement radical lorsqu’on les applique aux méthodes actuelles de prédiction de novo, les contacts prédits ne jouant visiblement qu’un rôle restreint dans le repliement [98].
2.3
Domaines d’application des couplages
évolutifs
L’arrivée d’une méthode de prédiction de contacts fiable ouvre un champ d’applica-tion relativement vaste qui s’étend en dehors des méthodes de prédicd’applica-tion structurales. Des outils de repliement de novo comme EVFold [76], pconsFold [95] ou plus ré-cemment CONFOLD [99] ont pu montrer l’efficacité de ce concept à retrouver in
silico des modèles proches des structures cristallographiques. Cette information peut
également être utilisée pour combler les lacunes des méthodes comparatives. Les données de contact peuvent par exemple faciliter la modélisation par homologie lorsque le modèle utilisé est relativement éloigné de la protéine cible1[100-102]. La prédiction des cartes de contacts peut également être utilisée pour reconnaître les classes de repliement [103, 104] et faciliter leur analyse [105]. La prédiction et l’étude de la dynamique des interfaces entre protéines [106-109] ou ligand [110-114], les méthodes de remplacement moléculaire en cristallographie [115, 116] ou encore les algorithmes d’alignements de structures [117, 118] figurent également parmi les applications possibles.
1. le principal indicateur étant une similarité de séquence en dessous de 30 %
3
Modélisation d’une structure par
Résonance Magnétique Nucléaire
Deuxième approche en terme de structure publiés, la détermination de structures par RMN permet d’explorer les états alternatifs d’un système [119]. Elle est constitue souvent la meilleur alternative dès lors qu’on rencontre des difficultés conséquentes en radiocristallographie [120]. Contrairement à cette dernière conduisant à la re-construction d’une carte de densité, l’interprétation des données RMN n’aboutit pas directement à un modèle structural mais à un ensemble de contraintes géométriques et nécessite obligatoirement de modéliser ces données expérimentales par l’inter-médiaire d’algorithmes de simulation moléculaire afin de générer un ensemble de conformères.
3.1
Données structurales en RMN
La principale information structurale apportée par les expérimentations en RMN apporte des données sur la distance entre les noyaux à partir des mesures des effets Overhauser. En fonction de l’intensité du pic associé à une paire de spins, la distance obtenue peut être de l’ordre de 1,8 Å jusqu’à 5 Å si l’intensité pour une intensité faible. Une approximation couramment acceptée est la formulation ISPA (Isolated Spin Pair Approximation) pour des temps de relaxation courts où l’intensité d’une mesure d’effet NOE est proportionnelle à r16 (r étant la distance
inter-atomique). Dans des milieux partiellement orientés, la portée des interactions dipolaires résiduelles (RDC)est plus importante permettant d’observer des distances au delà de 5 Å. L’intensité est dans ce cas proportionnelle àr13 mais dépend également
des angles θ entre les liaisons covalentes et le vecteur du champ magnétique statique. Cette valeur permet ainsi d’orienter les liaisons au cours de la modélisation.
Parmi les autres sources d’information, on peut entre autre ajouter des contraintes aux angles de torsions ϕ et χprovient des constantes de couplages scalaires observées entre les noyaux séparés par une ou plusieurs liaisons covalentes. La force est mesurée par les constantes de couplage qui dépendent du type des noyaux impliqués. La comparaison des valeurs observées dans les listes de déplacements chimiques avec celles provenant d’une pelote statistique permettent également de déduire de l’information supplémentaire sur les structures secondaires (hélice α et brin β). Cette approche se traduit par des contraintes d’angles dièdres générées habituellement par des outils dont le logiciel TALOS+ [121].
Chapitre 3 Modélisation d’une structure par Résonance
3.2
Détermination d’une structure à partir de
contraintes expérimentales RMN
Historiquement, construire un ensemble de modèles cohérents avec les données RMN s’appuit sur les algorithmes de géométrie de distance [122, 123]. Dans ces approches, on cherche à reconstruire les coordonnées atomiques internes [124-126] ou cartésiennes [127, 128] à partir des intervalles de distances associées aux me-sures d’effets Overhauser. Ces dernières ne conduisant pas nécessairement vers une solution réaliste, les protocoles se sont peu à peu orienté vers la combinaison avec une étape de dynamique moléculaire à partir de la structure générée par géométrie de distance [129, 130]. L’intérêt est ici de faciliter la phase de recherche en évitant de partir d’une configuration aléatoire. Au vu du temps de calcul important de l’algorithme et la performance croissante des modélisation, l’approche hybride a également laissé place ces dernières années au profit de méthodes purement méca-niques à partir d’une conformation aléatoire et utilisant des fonctions de pénalité associées aux données expérimentales.
3.3
ARIA : une approche itérative d’attribution de
données RMN et calcul de structure
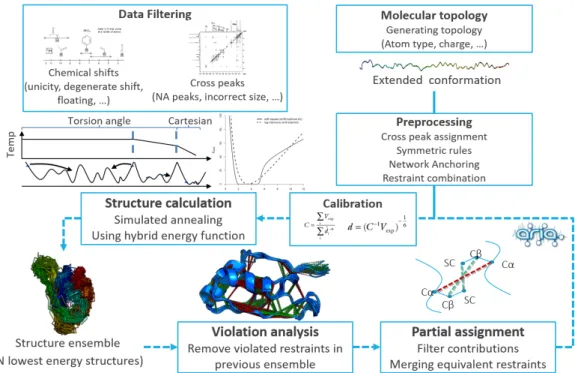
Une des principales difficultés en RMN pour résoudre une structure concerne l’attri-bution des données. En fonction du modèle étudié, il n’est pas rare de rencontrer des situations où les mesures restent relativement ambiguës. Afin d’éviter des er-reurs d’interprétations et faciliter la conception de structures, des méthodes comme l’approche ARIA [29-32] ont été mises en place.
3.3.1
Traitement itératif des données
Dans le protocole standard d’ARIA présenté dans la figure I.F, l’attribution des spectres extraits d’expérimentations en phase liquide ou solide est entièrement automatisée par un processus itératif (neuf itérations par défaut) permettant d’épurer tour à tour les erreurs d’attributions et les différentes possibilités dans le cas de données ambiguës en fonction des distances observées dans les modèles calculés.
Un calcul habituel d’ARIA commence par une étape de filtration des données no-tamment pour les tables de pics croisés (volume manquant ou négatif) les listes de déplacements chimiques (attribution spécifique entre un atome et une fréquence,
Figure I.F: Détermination de structure RMN à l’aide du protocole itératif d’ARIA
groupement prochiral1, attribution multiple). Une fois filtrée, ces listes seront utili-sées pour attribuer les pics croisés en fonction des tables de déplacements chimiques et générer un ensemble de contraintes de distances ambiguës ou non selon la précision des pics croisé et la tolérance choisie.
En parallèle à la génération de la topologie et d’une structure étendue aléatoire, plusieurs approches ont été implémentées pour traiter les listes de contraintes générées comme l’utilisation de règles empiriques pour classer les contraintes inter ou intra monomériques (en fonction de l’information de structure secondaire) dans le cas où on souhaite modéliser un oligomère. La méthode d’ancrage de réseau ou la combinaison de contraintes sont deux autres concepts dérivés des travaux d’Herrmann et al. [26] implémentés dans ARIA permettant d’affiner la liste.
Pendant le processus itératif, les distances dérivées des volumes des pics croisés sont calibrées en fonction des distances moyennes observées dans les n plus stables conformations calculées à l’itération précédente (n vaut en général 15 à 30% du nombre de structures générées). Ces distances ne pouvant tenir compte de l’ensemble des biais introduit (précision variable des appareils de mesure, transfert indirect d’aimantation, ...), elles sont habituellement interprétées dans la suite du procédé en intervalles de distances pour éviter toute distorsion du système due par exemple à la présence de bruit dans les données expérimentales.
1. groupement où la substitution d’un des atomes par un atome lourd donne un groupement chiral. En fonction de sa position, l’atome peut alors correspondre à un ou deux déplacements chimiques
Chapitre 3 Modélisation d’une structure par Résonance
Une solution adoptée dans ce protocole pour le traitement des pics sans attribution unique traite les données sous la forme de contraintes ambiguës. Elles partagent dès lors l’information avec plusieurs contributions sous la forme d’une distance effective (paires d’atomes possibles). Ce concept illustré dans la figureI.Gatraduit l’ensemble des distances associées aux différentes possibilités d’une contrainte ambiguë en une seule distance effective par la formule suivante :
D = Nc X c=1 d−6c !−16 (3.1)
Où dcet Nccorrespondent respectivement à la distance
a
b
Figure I.G: I.Ga: Illustration d’une contrainte de distance ambiguë (partie supérieure) ou
d’un ensemble de contraintes simples (partie inférieure).I.Gb: Potentiel cible
harmonique à fond plat (lignes pleines) ou log-harmonique (lignes pointillées)
L’analyse des contraintes se décompose en plusieurs parties. Pour chaque contrainte de distance durant l’étape de violation, la fraction de structures respectant la contrainte est calculée. On vérifie pour cela si la distance associée à cette der-nière est en dehors des limites fixées pour l’itération actuelle (dépendant du seuil dégressif de tolérance définit par l’utilisateur et des limites associées à la contrainte).