Maladie de Huntington
Texte intégral
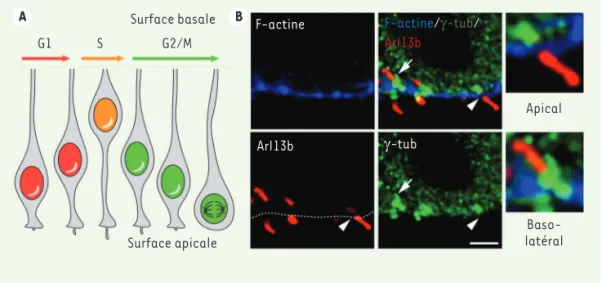
Figure
Documents relatifs
Les démarches de performance, engagées par un nom- bre croissant de collectivités locales depuis plus de trois ans, se caractérisent par l’implémentation de nouveaux principes,
Faire une longue récitation de Coran au cours de la prière du matin SOBH, une autre longue au cours de celle du DOHR, une courte pendant celle de ASR et celle de
Premier article avec une longue soie interne, le second avec deux soies apicales, dont l'externe est plus courte; exopodite triarticulé, avec 4 soies et épines
infectés à un jour d'âge, on observe une immunodépression beaucoup plus importante et plus longue. Sur le terrain, les poussins bénéficient généralement d'une protection maternelle
198 longue est fortement liée à la chromatine (voir Figure 20 C). La forme courte est beaucoup plus labile mais présente une affinité indéniable avec la chromatine.
La période courte est voisine dans les deux cas (5 jours), la période longue a une durée légèrement plus grande chez Mytilus galloprovincialis (130 jours) que chez Unio requieni
Demandez-leur : « Pensez-vous qu’il vous faut une ficelle plus longue ou plus courte pour que le pendule se balance 60 fois en une minute.. » Ils peuvent raccourcir leur fil de 50
La durée du voyage est plus courte selon les observateurs qui se déplacent dans une direction par rapport à Justine et elle est plus longue pour les observateurs qui
