Investigation of bound and unbound phosphoserine phosphatase conformations through Elastic Network Models and Molecular Dynamics simulations
Texte intégral
Figure
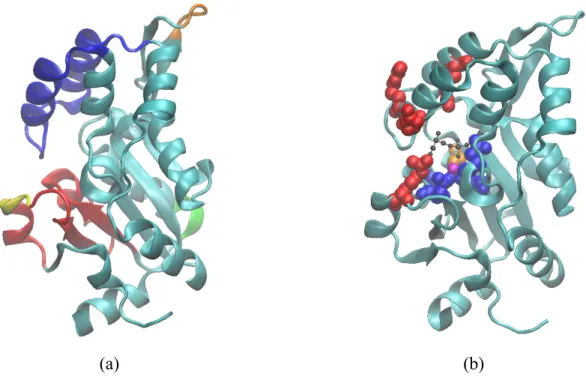
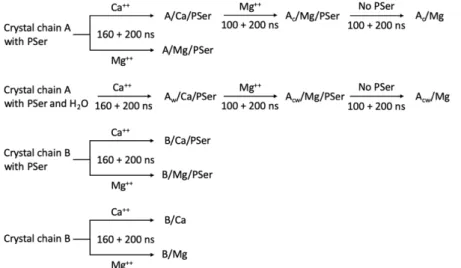
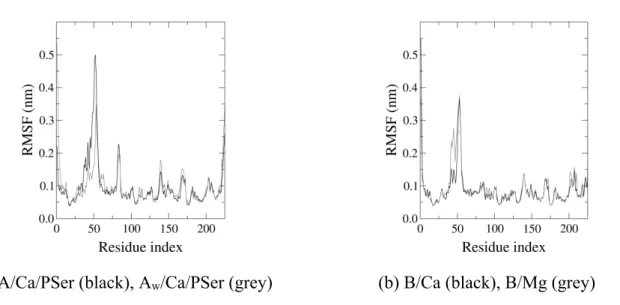
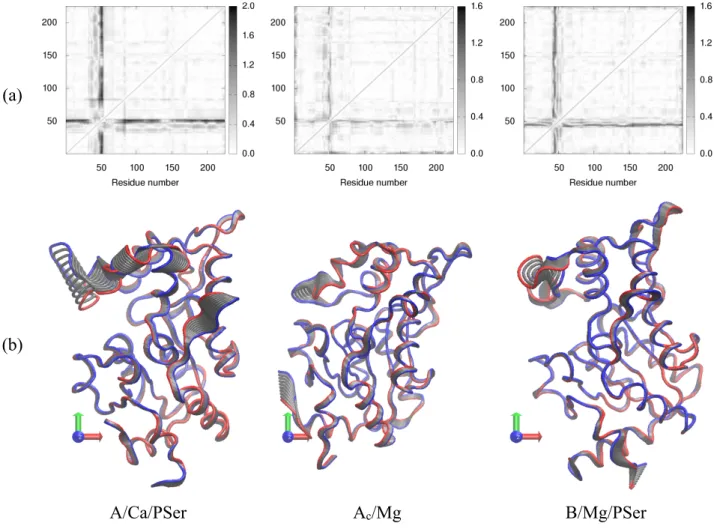
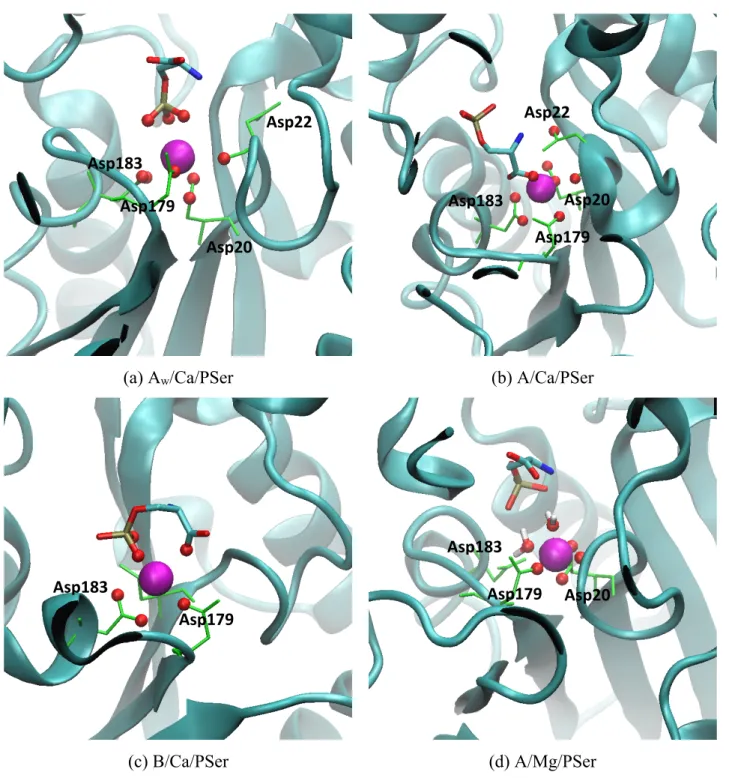
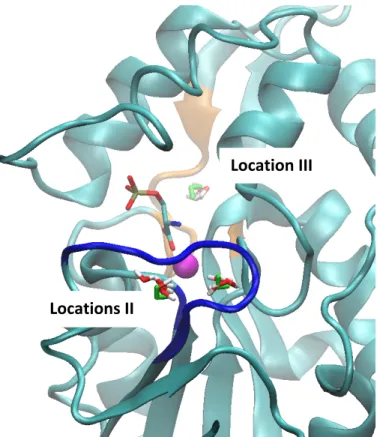
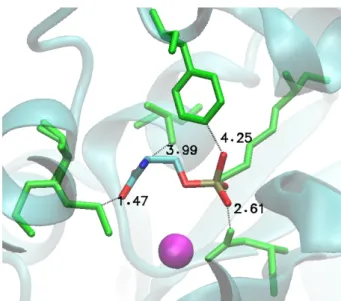
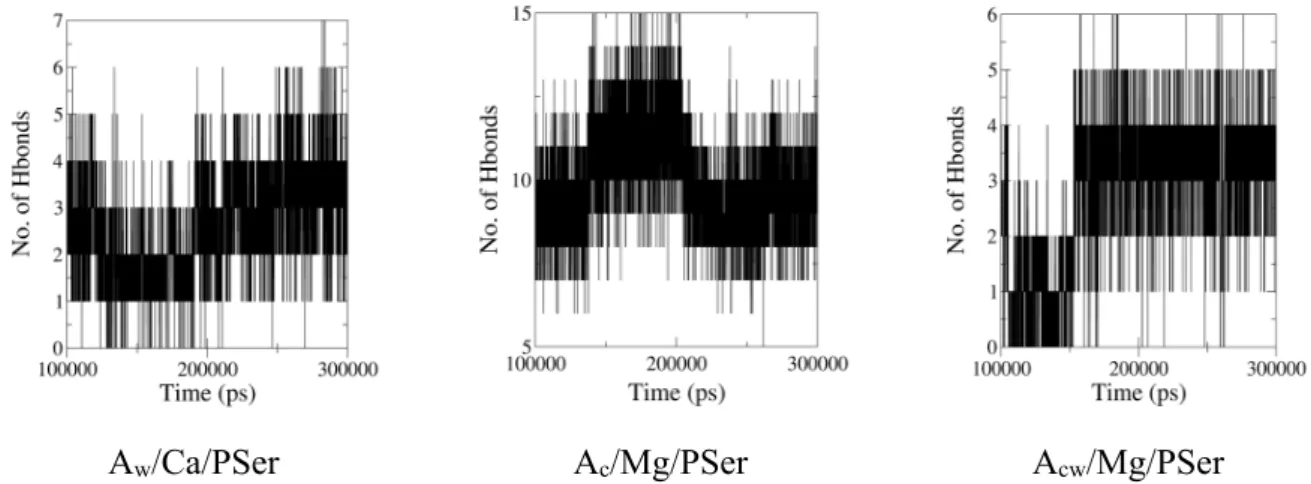
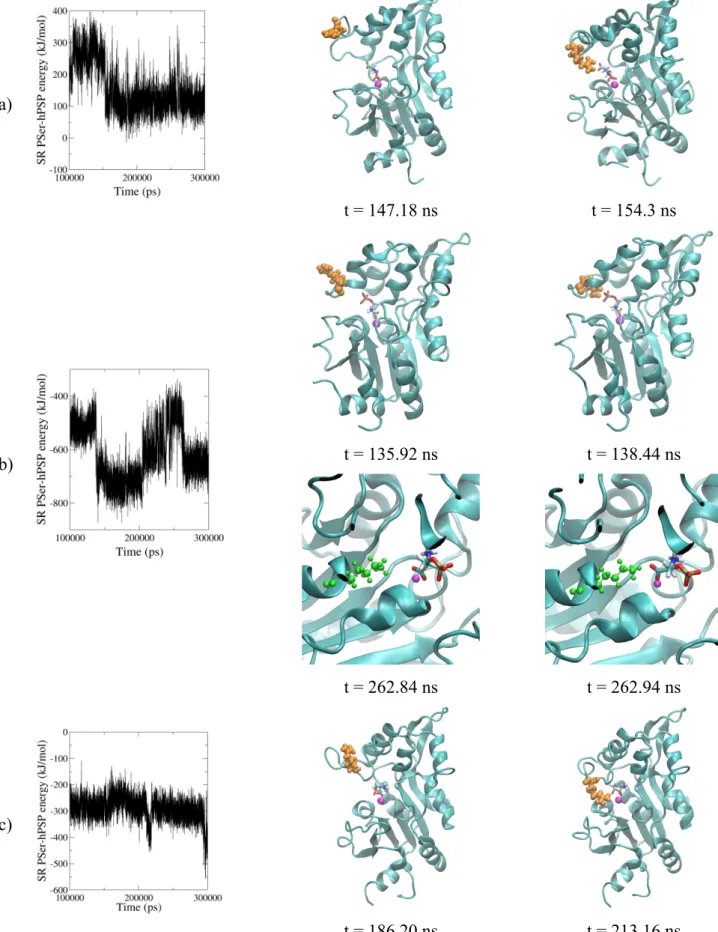
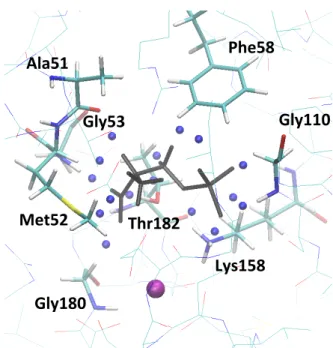
Documents relatifs
Elastic constants are calculated with comparison with experimental ones and then a represented amorphous hydrated calcium silicate-11Å tobermorite, obtained by
Table S2: Residues, atoms and corresponding distances and percentage of presence along the simulation time for the interface contacts predicted by the MD simulations for
We review recent results from computer simulation studies of polymer glasses, from chain dynamics around glass transition temperature T g to the mechanical be-.. haviour below
In termini concreti: nei programmi e nella politica la DSC fa più spesso riferimento ai diritti umani, integra più sistematicamente i principi inerenti ai diritti umani, pone in
In the present work, we assess the validity of elastic models for the shear viscosity and the α-relaxation time of supercooled water, using molecular dynamics simulations with
- Typical results obtained for the elastic response constant B versus temperature for the isometric series with 1 =
combination of rotation of the lipid molecule about the normal plus an out-of-plane diffusion inside a box (model I) and b) a superposition of in- and out-of-plane diffusion of
