HAL Id: hal-03162148
https://hal-amu.archives-ouvertes.fr/hal-03162148
Submitted on 9 Mar 2021HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Bernard-Soulier syndrome: first human case due to a
homozygous deletion of GP9 gene
Dorsaf Ghalloussi, Caroline Rousset-rouvière, Cornel Popovici, Florentine
Garaix, Noémie Saut, Paul Saultier, Michel Tsimaratos, Hervé Chambost,
Marie-Christine Alessi, Véronique Baccini
To cite this version:
Dorsaf Ghalloussi, Caroline Rousset-rouvière, Cornel Popovici, Florentine Garaix, Noémie Saut, et al.. Bernard-Soulier syndrome: first human case due to a homozygous deletion of GP9 gene. British Journal of Haematology, Wiley, 2020, 188 (6), �10.1111/bjh.16374�. �hal-03162148�
Correspondence
Bernard-Soulier syndrome: first human case
due ta a homozygous deletion of GP9 gene
A
9,
Dorsaf
Ghalloussi
1Caroline
Rousset-Rouvière
2Carnel
Popovici
3Florentine
Garaix
2Noémie
Sau
4Paul
Saultier
5Michel
Tsimaratos
2Hervé
Chambost
5Marie-Christine
Alessi
1Véronique
Baccini
1,�
Email veronique.baccini@chu-guadeloupe.fr AQ2 AQ31
UMR 1062 NORT, INSERM, Marseille:( France
2
Department of Multidisciplinary
.
Pêqtat:tics, Pediatric Nephrology Unit, La
Timone, University Hospital of JYlârseille, Marseille, France
3
Genetie Department, La TimoMê,-�niversity Hospital of Marseille,
Marseille, France
4
Hematology Laboratory, LàTtrnone University Hospital of Marseille,
Marseille, France
5
Department of Pediafr-iÇ H/èmatology, La Timone Hospital, Marseille,
France
Keywords
platelet ag�re9é3lipn; platelet disorders; platelet genetic diseases; platelet
me m bran e;J:hre>m bocyto pe ni a
Berna.rc:l-:$001.ier Syndrome (B55) is a rare (1 :1 million) hereditary bleeding disorder caus�q �tdèfects in the platelet glycoprotein (GP)-Ib/IX/V complex, a receptor for von Willebrand factor (VWF) and thrombin (Lanza, 2006; Bern dt & Andrews, 2011 ). Patients typitaUy present with epistaxis, petechial or gingival bleeding with onset already in infancy. They present with macrothrombocytopenia and their platelets do not àgglutinate in response to ristocetin, while maintaining a normal aggregation in r�sponse to a variety of aggregating agents. GPib/IX/V complex consists of two GPiba and four GPibl3 subunits stabilized by disulphide bonds (Luo et al., 2007). This heterodimer is non-covalently associated with two GPIX and one GPV subunits. The
N-terminal residues of GPiba form seven leucine-rich repeats (LRRs) and include the binding sites for VWF and thrombin. BSS is due to biallelic loss-of-function pathogen variants (deletions, insertions and nonsense mutations) in GPIBA, GPIBB or Ge9 genes encoding GPib/lX/V complex (Savoia et al., 2014). However, so far, no mut�tI(?� , • in GPS causing BSS has been reported yet. Most of the mutations prevent .the� formation of the complex or trafficking it through the endoplasmic reticulürniand Golgi apparatus and alter receptor expression (Salles et al., 2008; Savoia etial., 2011; Nurden et al., 2012).
The International Consortium for the study of BSS described 60 gen�Vé:lriations in
GP1 BA (28%), 59 in GPIBB (28%) and 92 in GP9 (44%) (Savoia et a/;, 2Q� 4). Most of
variations (85%) were homozygous and most cases were products<e>f/tonsanguineous
marriages (more than 50% of the families, mostly first cousins)./Familyrnembers with
only one mutant allele (BSS carriers) are usually asymptoma�i�/V)lith normal platelet
counts; however, they may sometimes show slightly enlarged plat�Jets and decreased GPib/V/IX complex expression, as well as a moderately requcedresponse to ristocetin
(Noris et al., 2012).
Here, we describe the first human case of BSS due todelètidn of the entire GP9 gene.
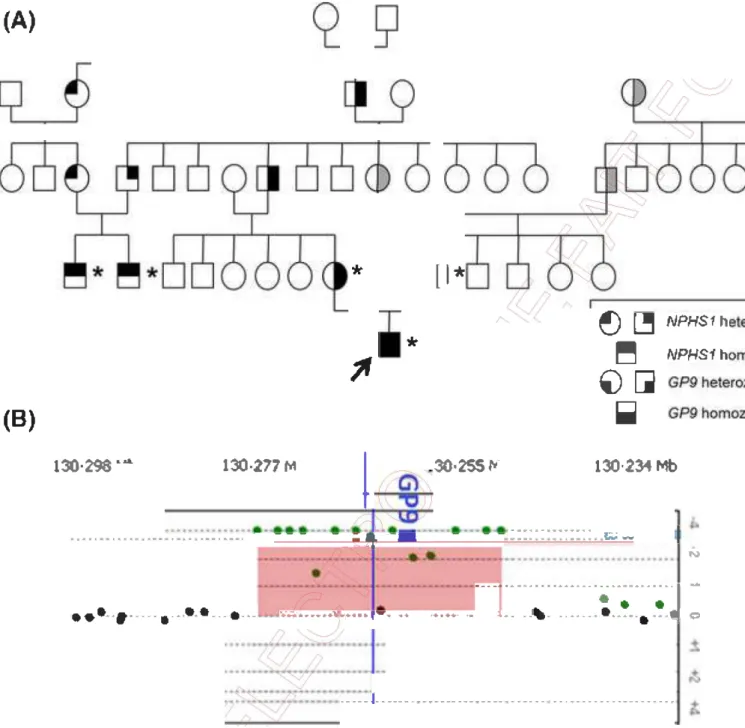
A male newborn developed a severe form of ���g�l"lital nephrotic syndrome of the Finnish type (CNS) with proteinuria, hypoalburnina�mia and oedemas. He is the first baby of a consanguineous couple. A homozyg�u:>mutation in the NPHS1 gene was previously identified in two other family r:nel)lb_ers and confirmed in our patient (Fig
--(A)
(B')
*
l
l
i
*
,,
1
·
w
{!) �
� NPHs1NPHS1ho·h ��IIli
l 30,• 298: "' .. , 130,277 t,{1
30.
255
,
r·
l30·234Mb-
....
.
...
1
••••�•••••••1•1•••••••••ir••••••• - - - - �·, �- -- - -•-•••••·•••�••·•••,._ll.zic il ... -,...
..
.
.
....
.
.
.
..
.
.
.
.
.
.
.
.
.. .
N···
·
·
··
···
- -···
·
�
-··
iil··
·
·
····
•J
1
-�W.---�-�•�-
.... ,.
..
... ..
,1,._ .J, ... ,. __ -� •�···-· .... -1 Q ... .. , ..• 11 .. ----•·-••· ... -... ,.,,..,. .. ----... --� -•.,. .. -... ,.. .... •111 - .. �Fig. 1 (A) Genealogictr�e; �• - genotype persans; grey - possible genotypes. (B) Screen shot frorrt=-afr_ay-CGH analysis showing the homozygous deletion of GP9gene.
AQS AQ6
The prôgnosjs for CNS is poor as the majority of cases die within six months of life. Howeyetji.r,travenous albumin supplementation, nutritional management, treatment of complications, dialysis and renal transplantation have been shown to improve the grovvth•and development of affected children (Holmberg et al., 1995).
TheNPHS1 gene has a size of 26 Kb and 29 exons. lt codes a transmembrane protein
{lamed 'nephrin'. The detection rate for this gene mutation varies among different ethnie groups. lt approaches 98% in Finnish children with CNS (Kestila et al., 1998).
During hospitalization, laboratory studies disclosed a profound thrombocytopenia
(9 g/1).
AQ7
Antibodies to HPASb were present in the maternai serum while the father's p.tatelèts
were HPASb-positive. So, the selected diagnosis was a maternai alloimmunizël.tion.
AQ8
Treatment for the CNS was initiated with daily intravenous albumin su<ppl�rr,�ntation,
thyroxine, antithrombin and intravenous immunoglobulin. Angioterî�in-tonverting enzyme inhibitors and non-steroidal anti-inflammatories dru.gs/ w�re added. Nephrectomy and renal transplantation were programmed.
However, two months later, the thrombocytopenia wa� p�r.sTstent with high transfusion frequencies (one per week). The morphol��y
of
the patient's bone marrow cells revealed the presence of some small <Œl�Qéll<aryocytes (MKs) with reduced cytoplasm and sometimes vacuolated (Fig 2AVand t>1 1 .. · .·J..... o .. od smear examinationrevealed giant platelets (Fig 2B).
(C) Cor:ikcl
-
�
O. :
I
_Fh�l•ki
�
,
1
..
..
l• "i· � � t,t �!W:ill �D411� iuo. pJll:i Gp,IXFig. 2il3o��
j-=
marrow smear (A) revealed the presence of some small
megakary9cytes with reduced cytoplasm and sometimes vacuolated and
bloqçlSrnêar (B) showed giant platelets (arrow). Flow cytometry labelling was
realit�p on platelet-rich plasma obtained after centrifugation for the contrai
and after whole-blood sedimentation for the patient because of the platelets'
s{Ze. Platelets' patient were negative for GpIX (C, right) like immunolabelling
çf Gplb/lX/V complex of patients' megakaryocytes (D, lower) whereas contrai
platelets (C, left) and megakaryocytes (D, upper) were positive.
In vitro study of patient's MKs derived from bone marrow co34+ cells in the presence
of thrombopoietin (TPO) and stem cell factor (SCF) (Bluteau et al., 2014) revealed IJO
differences in percentage of CD41 + ce lis and in the ploidy level (Lardier et al., 20J2) ',.
between the patient and an healthy donor (Figures S1 and S2).
Platelet glycoprotein surface expression was explored by flow cytometry and showed an absence of GPIX expression (Fig 2(). Likewise, immunolabelling of megc:1kà{y�cytes on a bone marrow smear was negative for GPiba showing the absence
of
GPib/IX/V complex expression (Fig 2D).Sanger sequencing of the patient's GPIBA and GPIBB did no(/reveaf damaging variation but the GP9 gene was not amplifiable. So, we analyzfi:?d thepNA extracted from peripheral whole blood by array-comparative genomic hx;br_içlizi3tion (array-CGH) by using the commercial Agile nt 1 M SurePrint G3 Hu man
<
�?1-:1
I\/lrcroarray (Agilent Technologies, Santa Clara, CA, USA) with an overall rl?!:!�ian/ and average probespacing of 2·6 and 3 Kb, respectively. The data wer�/e�vê;lcted using the Feature Extraction v.10.7.1.1 software (Agi lent Technologies),,ranalyz:ed with DNA Analytics
v.4.0.85 software (Agilent Technologies) using the �[)��2:9fgorithm with a threshold
of 5, and compared with the human genome referencE:! �equence hg19. The array-CGH analysis revealed a homozygous deletion on theTÇ)fl§ a-rtn of chromosome 3 (3q21.3) between the genomic positions 128 770 580 ëlliâ l28 796 076 (first and last deleted probe, hg19), so a deletion in the range 26-3\_ Kb çontaining only the GP9 gene (Fig
1 B). ' /
Four deletions encompassing the G�B(
ÇJEl�è are reported in the ClinVar database
(Landrum et al., 2014). Three of thesEic:1re very large deletions identified in individuals
with developmental disabilities ô( congenital anomalies. The fourth deletion, identified in a clinical setting, is dèsçrtbécl as a gross deletion encompassing the GP9 gene with unknown boundaries. Hc)W!:!\/er, the zygosity and clinical data are lacking.
Our report is the first hulTlàn_ Cë1se of homozygous deletion encompassing only the GP9 gene in a patient carrying/another genetic abnormality in a context of strong consanguinity.
Since diagnosis, hE:?_�ëlr oi-nephrectomized and kidney-transplanted for the CNS.
Platelets are no: :longer transfused and bleeding complications are treated by
tranexamic acici yvithout systematic platelet transfusion.
-Acknowledg�m,nts
X: Dorsë1f9ha-H�ùssi, Carnel Popovici, Noémie Saut and Véronique Baccini. Y: Marie Christine _ • .Ai.J!:!.ssi and Véronique Baccini. Z: Dorsaf Ghalloussi, Caroline Rousset RouVièDe/ .Fôrnel Popovici, Florentine Garaix, Noémie Saut, Paul Saultier, Michel Tsimaratqs, Hervé Chambost, Marie-Christine Alessi and Véronique Baccini. A: Dorsaf Ghc:1HolJssi, Carnel Popovici, Noémie Saut and Véronique Baccini. B: Véronique Baccini.
AQ9
ÂQlO
Fig S1. Megakaryocyte differentiation was deduced from control (A) or patient (B)
peripheral blood co34+ cells and analyzed at day 9 of culture.
AQ4
Fig S2. Megakaryocyte differentiation was deduced from control (A) or patient (B)
peripheral blood co34+ cells and analyzed at day 9 of culture.
References
Berndt, M.C. & Andrews, R.K. (2011) Bernard-Soulier syndro Haematologica, 96, 355-359.
Bluteau, D., Balduini, A., Balayn, N., Currao, M., Nurdert;p.,[.)eswarte, C., Leverger, G., Noris, P., Perrotta, S., Solary, E., Vainche��et))W., Debili, N.,
Favier, R. & Raslova, H. (2014) Thrombocytopenia�a�qdated mutations in the ANKRD26 regulatory reg ion induce MAPK hyperaoJivatinn. Journal of Clinical Investigation, 124, 580-591.
Holmberg, C., Antikainen, M., Ronnholm, �-,��-H-ouhala, M. &Jalanko, H. (1995) Management of congenital nephrotit syndrome of the Finnish type. Pediatric Nephrology (Berlin, Germany), �' �?:-�3.
Kestila, M., Lenkkeri, U., Mannikko,/M.;��rnerdin,
J.,
McCready, P., Putaala, H., Ruotsalainen, V., Morita, T., Nissir-tert/J\A., Herva, R., Kashtan, C.E., Peltonen, L., Holmberg, C., Olsen, A. & Tryggyaso�, K. (1998) Positionally cloned gene for a novel glomerular protein-nephrI�-is; mutated in congenital nephroticsyndrome. Molecular Cell, 1.1575""582.
Landrum, M.J., Lee, J.M.,<�ll:Y·.G·R., Jang, W., Rubinstein, W.S., Church, D.M. &
Maglott, D.R. (2014) CUnVar: public archive of relationships among sequence variation and human ph�np.type. Nucleic Acids Research, 42, D980-D985. Lanza, F. (2006) B-ey,�r:_d-Soulier syndrome (hemorrhagiparous thrombocytic dystrophy). Orphéln�t Journal of Rare Diseases, 1, 46.
Lardier, L., Bll.l't:eau, D., Jalil, A., Legrand, C., Pan,
J.,
Rameau, P., Jou ni, D.,Bluteau, ç;>:,>Met�her, T., Leon, C., Gachet, C., Debili, N., Vainchenker, W.,
Raslova�kL�.Chang, Y. (2012) RUNX1-induced silencing of non-muscle myosin
heavrch;�Ln_IIB contributes to megakaryocyte polyploidization. Nature CommL.JQic-ations, 3, 717.
Luo,
S.Z., Mo, X., Afshar-Kharghan, V., Srinivasan, S., Lopez, J.A. & Li, R. (2007) <3Iyf�protein Ibalpha forms disulfide bonds with 2 glycoprotein Ibbetasubünits in the resting platelet. Blood, 109, 603-609.
oris, P., Perrotta, S., Bottega, R., Pecci, A., Melazzini, F., Civaschi, E., Russo, S., Magrin, S., Loffredo, G., Di Salvo, V., Russo, G., Casale, M., De Rocco, D.,
Grignani, C., Cattaneo, M., Baronci, C., Dragani, A., Albano, V., Jankovic, M., Scianguetta, S., Savoia, A. & Balduini, C.L. (2012) Clinical and laboratory