Surface sterilization methods impact measures of internal microbial diversity in ticks
Texte intégral
Figure
Documents relatifs
Despite clear differences in the microbial communities of different tick genera (Figures 2 and 3), several bacteria genera were shared by all tick genera including
The objective of our study was to identify the infection and co-infection rates of different Borrelia genospecies along with other tick-borne pathogens in questing ticks collected
In promoting heteroge- neous, translingual language spaces and practicing difference as a form of linguistic dissidence, artists such as the Breton singer Erik Marchand and the hip
Title: Genetic diversity of Ehrlichia ruminantium in Amblyomma variegatum ticks and small ruminants in The Gambia determined by restriction fragment profile analysis Authors:
This study focuses on biofilm microbial communities in a trop- ical mangrove with two different trophic statuses, either exposed or not exposed to effluents from a domestic
Despite the difference of sensitivity of the different algal species to GBMs, the concentrations leading to 50% of algal growth inhibitions all ranged from 20 to over 150 mg.L −1
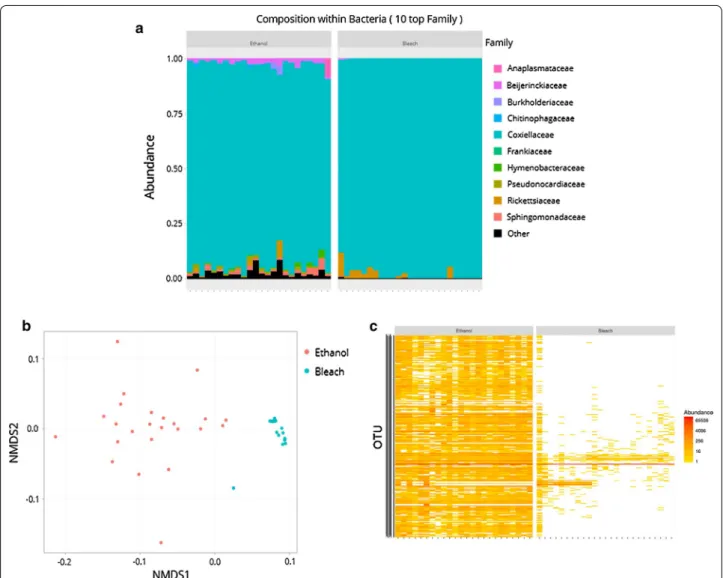
further observations are particularly relevant: (i) there was no difference in bacterial diversity between etha- nol- and bleach-treated ticks, showing that surface steri-
ruminantium isolate structure in Mozambique, cattle and wildlife were sampled across the south and center of Mozambique as well as in the adjacent Kruger National Park (KNP),
