DNA Methylation Detection using an Engineered
Methyl-CpG-Binding Protein
by
Brooke Elizabeth Tam
B.S. Chemical Engineering
The Ohio State University, 2012
Submitted to the Department of Chemical Engineering
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy in Chemical Engineering
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
June 2018
2018 Massachusetts Institute of Technology
All rights reserved.
Signature of A uthor...
Brooke Elizabeth Tam
Department of Chemical Engineering
May 9, 2018
Signature redacted
C e rtified by...
Hadley D. Sikes
Associate Professor of Chemical Engineering
Thesis Supervisor
Accepted by
... ...
S ig n atu re red acted
Patrick S. Doyle
-N
Professor of Chemical Engineering
MAY
- 12019
Graduate Officer
DNA Methylation Detection using an Engineered Methyl-CpG-Binding Protein by
Brooke Elizabeth Tam
Submitted to the Department of Chemical Engineering on May 9, 2018 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Chemical Engineering
Abstract
DNA methylation, specifically the methylation of cytosine bases, is an important biomarker, as
abnormal DNA methylation patterns are found in many different types of cancer. Currently, a small number of cancer hospitals evaluate the methylation status of the MGMT gene promoter to determine the best course of treatment for patients with glioblastoma. However, improved methylation detection techniques are required in order to expand the availability of such testing to more patients.
Methyl-CpG-binding domain (MBD) proteins bind specifically to methylated DNA sequences, and many assays have been developed that use these proteins in methylation profiling of DNA. The wild-type proteins in the MBD family bind specifically to symmetrically methylated CpG dinucleotides. Here, I have engineered a new MBD variant that binds to hemi-methylated DNA but not unmethylated DNA, allowing for the detection of a methylated target sequence
hybridized to a simple, unmethylated DNA probe. With four amino acid substitutions, a protein that did not show any binding to hemi-methylated DNA at concentrations up to 100 nM was altered to bind hemi-methylated DNA with high affinity. Based on equilibrium binding titrations, this engineered variant binds a DNA sequence with a single hemi-methylated CpG dinucleotide with a dissociation constant of 5.6 1.4 nM.
After engineering a protein to bind hemi-methylated CpG dinucleotides, I developed a simple, hybridization-based assay to determine the methylation status of the MGMT promoter using this protein variant and magnetic microparticles. The target DNA molecules are captured on the surface of magnetic microparticles and an MBD-GFP fusion protein is added to bind if the captured target is methylated. Therefore, MBD binding can be detected directly based on fluorescence of the microparticles after the binding step without requiring any chemical conversion or additional labeling steps. In addition to simplifying the assay and eliminating the need for methylated capture probes, I was able to improve the sensitivity of the assay to 5 pM target DNA. Finally, I also studied the DNA capture and MBD binding events to identify the key parameters and guide future efforts to develop clinically relevant diagnostics.
Thesis Supervisor: Hadley D. Sikes
Contents
A b stra ct ... 3
List of Figures...7
List of Tables...8
Chapter 1 Introduction...9
1.1 Epigenetics and Precision M edicine ... 10
1.2 Promoter M ethylation as a Biomarker... 10
1.3 Current Practices for DNA M ethylation Analysis ... 12
1.4 New Technologies for DNA M ethylation Analysis ... 13
1.4.1 Emerging Bisulfite-based M ethods... 13
1.4.2 Bisulfite-free, PCR-based M ethods... 15
1.4.3 PCR-free, Affinity Agent-based M ethods ... 15
1.4.4 M ethylation Assessment in Fixed Cells ... 17
1.5 Clinical Outlook ... 18
1.6 Thesis Organization ... 19
Chapter 2 Characterization and directed evolution of a methyl-binding domain protein for high-sensitivity DNA methylation analysis ... 21
A b stra ct ... 2 2 2.1 Introduction... 23
2.2 M aterials and M ethods... 24
2.2.1 Displaying M BD proteins on the surface of S.cerevisiae yeast cells... 24
2.2.2 Characterizing MBD binding to DNA oligonucleotides with varying methylation patterns... 25
2.23 hM BD2 library creation using epPCR... 26
2.2.4 Library screening for MBD2 variants with improved binding affinity to methylated CpGs... 26
2.2.5 M easuring thermal stability of wild-type and evolved M BIs... 27
2.2.6 Bacterial expression of M BD2 variant proteins ... 27
2.2.7 Biochip-binding experiments and affinity determination ... 28
2.3 Results an d Discussion ... 29
2.3.1 Yeast surface display and characterization of M BD proteins... 29
2.3.3 Structural modeling for MBD variants with improved binding affinity to meDNA ... 37
2.3.4 Affinity enhancement by concatenation for interfacial binding applications...39
2.4 Conclusions... 41
Chapter 3 Engineering affinity agents for the detection of hemi-methylated CpG sites in DNA...42
Abstract ... 43
3.1 Introduction...44
3.2 M ethods... 45
3.2.1 Characterization of Binding Affinities using Yeast Surface Display... 45
3.2.2 Screening the MBD Library for Improved Affinity for Hemi-Methylated DNA ... 46
3.2.3 Soluble Protein Expression ... 47
3.2.4 Biochip Experim ents... 47
3.3 Results and discussion... 48
3.4 Conclusions... 55
Chapter 4 An Examination of Critical Parameters in Hybridization-Based Epigenotyping using Magnetic M icroparticles... 57
Abstract ... 58
4.1 Introduction...59
4.2 M aterials and M ethods... 62
4.2.1 Isolation of Genom ic DNA... 62
4.2.2 Probe/Target Hybridization and Capture on M agnetic Beads ... 62
4.2.3 M ethylation Profiling w ith M BD-GFP ... 62
4.3 Results a nd Discussion ... 63
4.4 Conclusions... 70
Chapter 5 Quantitative Evaluation of Protein Binding Properties and Usefulness in Sequence-Specific M ethylation Assays ... 72
Abstract ... 73
5.1 Introduction...74
5.2 M ethods...75
5.2.1 M odeling...75
5.2.2 Experim ental Target Hybridization Studies... 75
5.3 Results a nd Discussion ... 76
5.3.1 Target DNA Capture by Hybridization ... 76
5.3.2 M ethylation Detection by M BD Binding: Equilibrium -Based ... 81
5.4 Conclusions...89
Chapter 6 Conclusions and Future Directions ... 91
6.1 Conclusions... 92
6.1.1 Engineering a Protein to Bind Hem i-M ethylated DNA ... 92
6.1.2 Examining the Parameters in Hybridization-Based Epigenotyping and Developing a Bead-Based M ethylation Assay... 93
6.1.3 M odeling the Kinetics of M BD Binding... 93
6.2 Future Directions... 94
6.2.1 Engineering DNA Binding Proteins ... 94
6.2.2 Hybridization-Based Epigenotyping for Clinical Sam ples ... 95
List of Figures
Figure 1.1 A schematic demonstrating the general workflow once a glioblastoma sample is obtained ... 11
Figure 1.2 A schematic of the QuARTS assay performed on bisulfite treated DNA ... 14
Figure 1.3 A schematic of methylation detection based on the change in photoelectrochemical response u po n M B D binding ... 16
Figure 1.4 A diagram of a biochip assay with radical photopolymerization...17
Figure 1.5 A diagram of the M eFISH protocol ... 18
Figure 2.1 Synthetic DNA oligonucleotides derived from the MGMT gene ... 25
Figure 2.2 Yeast surface display of M BD family proteins ... 30
Figure 2.3 Detection and quantification of methylated DNA binding to yeast displayed MBD proteins .. 31
Figure 2.4 A reduced resolution, five-point equilibrium binding experiment, round 1...34
Figure 2.5 Sequence comparison of MBD containing proteins ... 35
Figure 2.6 A reduced resolution, five-point equilibrium binding experiment, round 2...36
Figure 2.7 The fraction of binding activity retained by MBD2 proteins after exposure to 70"C...37
Figure 2.8 Structural analysis of amino acid substitutions in MBD variant 2/5 ... 38
Figure 2.9 NxMBD2-Var2/5-GFP proteins bind to surface-immobilized DNA ... 40
Figure 2.10 Modeling MBD binding based on the dissociation constant... 41
Figure 3.1 Wild-Type M BDs binding to hemi-methylated DNA ... 49
Figure 3.2 Screening for clones with improved binding to hemi-methylated DNA... 50
Figure 3.3 Binding improvements obtained during library screening ... 51
Figure 3.4 A reduced resolution comparison of isolated clones. ... 52
Figure 3.5 An engineered MBD binds hemi-methylated DNA with high affinity...53
Figure 3.6 Structure of hM BD2 Variant H4... 54
Figure 3.7 A biochip assay w ith hM BD2 Variant H4 ... 55
Figure 4.1 Hemi-methylated DNA and hMBD2 Variant H4-GFP-B ... 61
Figure 4.2 Capturing target DNA on the surface of magnetic microparticles ... 64
Figure 4.3 Pre-hybridization of probe and target... 66
Figure 4.4 Optimization of hybridization times to improve target capture ... 65
Figure 4.5 A methylation assay on magnetic microparticles... 68
Figure 4.6 Performing the methylation assay in the presence of genomic DNA ... 69
Figure 5.1 Fraction of capture sites bound at varying target DNA concentrations...78
Figure 5.2 Bead density and the number of target molecules per bead...80
Figure 5.3 A model of M BD binding events at equilibrium ... 83
Figure 5.4 A m odel of an equilibrium binding assay... 84
Figure 5.5 Association and dissociation profiles for MBD binding ... 86
Figure 5.6 A kinetics-based assay when non-specific association and dissociation are fast...88
List of Tables
Table 2.1 Equilibrium dissociation constants for wild-type hMBD2, variant 1/4 and variant 2/5 binding to
DNA w ith one m ethylated CpG site ... 32
Table 2.2 Mutations to hMBD2 and the frequency observed during rounds 1 and 2 of MBD directed evolution by epPCR and flow cytometry screening of the yeast surface display library... 33 Table 2.3 Sequences of MBD variants from round 1 of error-prone PCR and screening...33 Table 2.4 Sequences of MBD variants from round 2 of error-prone PCR and screening...36 Table 3.1 Sequences of MBD variants isolated from the error-prone PCR library that was enriched for binding to hem i-m ethylated DNA ... 51
Chapter 1
Introduction
1.1 Epigenetics and Precision Medicine
Precision medicine promises to transform the way we treat patients with complex diseases such as cancer. Accurate profiling of both genetic mutations and epigenetic modifications that alter gene function or expression are critical to its success. One such epigenetic modification is cytosine methylation (5mC) where a methyl group is added to a cytosine base that is usually in the context of a CpG dinucleotide and thus symmetric on the antisense strand. This CpG methylation is typically a mechanism of gene silencing and is propagated through DNA
replication. Many studies have shown that abnormal DNA methylation is involved in cancer pathogenesis, and translational research efforts have begun to identify hypermethylated genes that can be used as biomarkers.1 To date, there is evidence for hypermethylation for more than forty-eight genes across tumors that arise in eighteen unique tissues. Nine of these
hypermethylated genes are measured as part of commercially available assays.2 Measurement of DNA methylation biomarkers has been applied to aid in cancer screening, diagnosis,
prognostic stratification, therapy assignment, and recurrence identification.' So far, two DNA methylation assays are accepted as standard of care in patients with glioblastoma and
colorectal cancer, and these two assays are routinely performed in some US cancer hospitals.1 As data on clinical utility of many other methylation biomarkers accumulates, assays of DNA
methylation will likely become more commonplace in the clinical molecular diagnostic
laboratory. A large part of the success of this class of biomarkers depends on the development of reliable and rapid analysis methods that are suited to clinical use.
1.2 Promoter Methylation as a Biomarker
Currently, the most widely tested cancer-related epigenetic biomarker is hypermethylation of the 06-methylguanine-DNA methyltransferase (MGMT) promoter, which predicts glioblastoma
response to alkylating chemotherapeutic agents.3 A diagram showing how MGMT methylation
assessment fits into the workflow of glioblastoma sample analysis is shown in Figure 1.1.
0. - 6 . - i
---Obtain biopsy e- omlnfx and MoeuarMM pooe
sample emnbed in paraffindagsts
Figure 1.1 A schematic demonstrating the general workflow once a glioblastoma sample is obtained. For
other cancers, the number and type of molecular diagnostic tests routinely performed may differ. The
most clinically feasible diagnostic tests should be compatible with a small section of formalin-fixed tissue.
The second accepted application of DNA methylation assessment is for MutL homolog 1 (MLH1) hypermethylation in a subset of colorectal cancers (CRCs) that demonstrate microsatellite instability and loss of MLH1 protein expression by immunohistochemistry. MLH1 promoter methylation is one factor in distinguishing sporadically occurring tumors from those occurring
as part of an inherited cancer syndrome with microsatellite instability.4-6 Many genes
recurrently mutated in sporadic or inherited cancer syndromes such as MLH1 are also subject to silencing by promoter methylation.7 Another important example is methylation of the BRCA1
promoter in breast and ovarian cancers. Olaparib, a poly(adenosine diphosphate)-ribose
polymerase (PARP) inhibitor, was recently approved by the FDA for treatment of BRCA-mutated
ovarian cancers. Human cancer models have demonstrated similar response to PARP inhibitors
for mutated and hypermethylated BRCA1,8 suggesting that additional patients could potentially
benefit from Olaparib treatment. As more targeted cancer therapies are approved, the number of hypermethylated genes predictive of treatment sensitivity is expanding.
In addition to analysis of surgical pathology samples, DNA methylation shows promise in cancer screening strategies. Recently approved by the FDA, the Cologuard' test generates a composite score for colorectal cancer (CRC) risk that includes promoter methylation analysis of the N-myc downstream-regulated gene 4 (NDRG4) and the bone morphogenetic protein 3 (BMP3), from a
stool sample.9 In a large clinical study, Cologuard* outperformed another accepted form of
stool-based CRC called fecal immunochemical testing with a sensitivity of 92% for cancer and specificity of 86-99% for both cancerous and pre-cancerous lesions relative to findings on
colonoscopy.9 Another assay, the Epi ProColon test, detects methylated Septin9 (SEPT9) DNA
from plasma with performance comparable to stool-based screening methods: sensitivity for all
colonoscopy findings of 80.0% (78%-82%).101 Additionally, methylation of the short stature homeobox 2 (SHOX2) gene is being investigated in plasma for monitoring treatment of
non-small cell lung cancer and in bronchial aspirates for aiding in diagnosis.12 in the field of
prostate cancer, multiplex analysis appears to be especially valuable. Measuring DNA methylation levels for a combination of the GSTP1, APC, and PTGS1 gene promoters can distinguish prostate cancer from benign hyperplasia and correlate with some prognostic features;14 however, to date, such an assay has not entered clinical practice. These screening
methylation assays are approximating but have not surpassed standard methods in their clinical performance and low cost. Further research to elucidate clinically actionable methylation sites as well as improvement in 5mc detection technology will hopefully expand the application of
DNA methylation measurement in medicine.
1.3 Current Practices for DNA Methylation Analysis
The widely used technologies for DNA methylation analysis can be coarsely divided into two categories on the basis of sequence coverage: genome-scale and one to several CpG sites. The ends of this spectrum represent a trade-off between quantity of information and cost. The genome-scale methods have been well studied and compared with each other;15
,16 these are the defacto standard for generating reference methylome data and enhancing our knowledge
of biology and disease. While critically important for discovery purposes, these methods are generally not well suited for routine analysis of clinical samples. In the clinical context, only one to several areas of interest may provide actionable information;' therefore, generating
genome-level data is an inefficient use of healthcare resources. For example, person-hours, expertise, instrumentation, and funds to devote to each analysis are more limited in the clinic. In addition, the substrate for clinical methylation analysis is often different from research material; samples are often formalin-fixed rather than fresh, sample size is limiting in many cases, and the tolerance for repeating failed analyses is lower.
One major technological challenge to developing clinical methylation assays is the
heterogenous nature of clinical samples. Surgical specimens are a complex mixture of normal and cancerous tissue, and there can even be extensive molecular heterogeneity within one tumor.2" 7 Therefore, assays must be very sensitive and specific when DNA from normal tissue
may outnumber DNA for diseased cells by many orders of magnitude. This is especially relevant to screening assays such as those using plasma and stool. Preservation of tissue with formalin fixation poses additional challenge as this may lead to DNA degradation, thus requiring optimization of assays to clinical samples.2
We surveyed seven of the US hospitals identified as providing the best cancer care by U.S. News
& World Report. Two of these hospitals, at the time of this writing, do not currently offer any DNA methylation testing in their pathology laboratories. The remaining five institutions all use
some form of methylation-specific PCR (MSP) for routine analysis.18 Two also report using massively parallel sequencing-based methods on a limited basis for clinical, non-research cases.
Methylation-specific PCR and sequencing based methods rely on bisulfite conversion of unmethylated cytosine bases to uracil by deamination. Chemical conversion alone, however, can degrade more than 90% of the sample DNA.19 20 Such losses incurred during DNA extraction and processing necessitate larger sample inputs on the order of hundreds of nanograms to micrograms of DNA.1 Many sample types lack this amount of DNA, such as blood plasma and body fluid specimens2 2
or samples with very little resected or biopsied tissue. Further, bisulfite conversion protocols must be rigorously optimized to maximize the conversion of
unmethylated cytosine to uracil and minimize improper reaction of 5-methylcytosine to thymine.2 3 Such conversion errors deleteriously affect the fidelity of results. Therefore, every
conversion reaction must also be checked by testing unmethylated DNA from HCT116 cells deficient for DNMT1 and DNMT3B as well as enzymatically methylated DNA as negative and positive controls, respectively.24 Consequently, it should not be surprising that four of the five surveyed institutions currently testing for hypermethylation identified the bisulfite conversion step as an aspect of the method they would most like to change or eliminate.
1.4 New Technologies for DNA Methylation Analysis
A handful of approaches have been devised to address the engineering challenges facing DNA
methylation analysis. The first strategy is to mitigate the deleterious effects caused by bisulfite conversion and amplify the remaining signal to provide sensitive detection of DNA methylation. Other strategies leverage PCR to amplify unconverted DNA prior to methylation analysis or detect methylation using an affinity agent. Many new methods in each of these categories have recently been developed, and each of these methods improve upon some of the problems associated with traditional methylation-specific PCR.
1.4.1 Emerging Bisulfite-based Methods
Many new methods are able to provide quantitative results with less input DNA than traditional methylation-specific PCR, which generally requires approximately 1 ig of DNA. One of these methods, Methyl-BEAMing, involves two sequential PCR reactions, one analogous to
methylation-specific PCR and a second that involves single-molecule PCR on a microparticle in an oil/water emulsion.2
s Flow cytometry or sequencing can then be used to quantify the number of methylated DNA fragments. While demonstrated using DNA from plasma and stool samples, the specialized bisulfite conversion protocol and lack of multiplex analysis make its clinical applicability uncertain. A similar approach, MS-qFRET, consists of a MSP reaction with forward and reverse primers functionalized with biotin and a fluorophore, respectively.26 The fluorescently labelled PCR products and a streptavidin coated quantum dot (QD) form a FRET pair when bound together and indicate DNA methylation with a change in the QD emission spectrum. Bailey and colleagues were able to detect 15 pg of methylated DNA in the presence of 150 ng unmethylated DNA.26They demonstrated that the method is sensitive enough to
detect methylated DNA in sputum samples. Additionally, the procedure integrates well with traditional equipment and off-the-shelf reagents. In another FRET-based approach, the
quantitative allele-specific real-time target and signal amplification (QuARTS)
QuARTS (Duplex Format)
Target 1Invasive oligo ctevage
Forward Primer 5 rget 1Probe
3,.
5'
Reverse Primer
Target 2
nasive oligo Cleavage
Forward Primer Target 2 Probe
Reverse Primer
Quencher v
Red Dye FRET Cassette
Reaction 3
=C==
Quencher Cleavage
FAM Dye FRET Cassette
4
- Fluorescent Signal EmittedFigure 1.2 A schematic demonstrating the DNA amplification and signal generation steps of the QuARTS
assay performed on bisulfite treated DNA.2
(Reproduced from Ref. 27 with permission from the American Association for Clinical Chemistry)
method, depicted in Figure 1.2, uses a gene-specific oligonucleotide probe to recognize a MSP amplicon which, through two sequential 5'-flap endonuclease reactions, liberates a fluorescent
dye that can be detected in a qPCR-like format.27 Not only does this approach provide
significant signal amplification, it can accommodate multiplexed detection of three separate genes in a single PCR reaction for a clinically relevant sample.
Other recently developed methods include single-cell whole-genome bisulfite sequencing
(scWGBS)28 and bisulfite treatment and detection within a microfluidic device.29
In scWGBS,
DNA degradation from bisulfite treatment does reduce genome coverage; however, aggregating low-coverage data from several single-cell analyses has been shown to provide more complete information. While best suited for research applications at this point, this technology has the potential to greatly impact basic research and hypermethylome biomarker
discovery. The single-channel microfluidic device developed by Yoon et aL.29 combines bisulfite
treatment with isothermal solid-phase amplification/detection. This method not only offers
14
better sensitivity than current methods, but it also reduces the total time required for bisulfite treatment and detection to only 80 minutes.
Another exciting innovation is a method that gives a colorimetric readout that can be seen with the unaided eye. This method, reported by Su et al.30
, utilizes ligase chain reaction after
bisulfite treatment to amplify the unconverted (methylated) DNA. Gold nanoparticles
functionalized with two different DNA probes, each complementary to a different segment of the target DNA, bind the amplified DNA in a sandwich hybridization. This leads to aggregation of the nanoparticles and a visible color change in the solution.30
Other readout methods utilizing chemiluminescence,3
1 fluorescence polarization,32 and surface-enhanced Raman scattering33
have also been developed.
1.4.2 Bisulfite-free, PCR-based Methods
While many improved methods have made bisulfite treatment and the subsequent detection steps easier, faster, and more sensitive, DNA degradation is still a limitation. Enzymatic steps such as PCR amplification have been combined with various methylation detection methods to eliminate the need for bisulfite treatment. Single-molecule, real-time (SMRT) sequencing measures the change in DNA synthesis kinetics that arise when the translating polymerase encounters modified nucleotides in the template strand. Primer design can be used to target the region of interest; however, SMRT is not capable of providing single-base resolution of 5mC. Conversely, others have described using an Oxford Nanopore to identify unlabelled nucleoside 5'-monophosphates, including 5mC, that could potentially be liberated from a single DNA strand as they are cleaved sequentially by an exonuclease to affect methylation-specific sequencing.3
s
Qiagen introduced the EpiTectg Methyl 11 PCR Assay, which stems from earlier methods developed to measure 5mC using restriction enzymes (e.g. Hpall and McrBC) that cleave differentially in response to DNA methylation states.3
' Digestion with a combination of methylation-sensitive and methylation-dependent enzymes followed by real-time PCR provides quantitative DNA methylation profiling. No quantitative evaluation of this method has yet been published in the peer-reviewed literature; however, the manufacturer claims a 2 Ig sample of genomic DNA is enough to interrogate up to 94 genes out of >37,000 promoter CpG islands for which PCR primers can be synthesized.
1.4.3 PCR-free, Affinity Agent-based Methods
There is a great potential to reduce assay complexity by eliminating the need for both bisulfite conversion and PCR amplification of target DNA. These new technologies generally rely on a protein affinity agent, either a methyl binding domain (MBD) or antibody, for specific recognition of 5mC; however, each differs significantly in detection mechanism. Label-free, optical biosensors use MBDs or antibodies immobilized on an opto-fluidic ring resonator3 8 or a
silica optical microtoroid resonant cavity3 9 to capture DNA and provide specific detection of 5mC and 5hmC (a hydroxylated 5mC variant that may represent a demethylation intermediate),
respectively. Currently, these methods are neither capable of providing sequence-specific detection nor have they been demonstrated using clinically relevant samples. However, Suter et al.38 mentioned addressing both of these by incorporating sequence-specific DNA probes and
testing samples from blood as future efforts.
In another method, MBD binding to 5mC in single DNA molecules with fluorescence detection
has been demonstrated in a high throughput nanofluidic device.40 This method offers
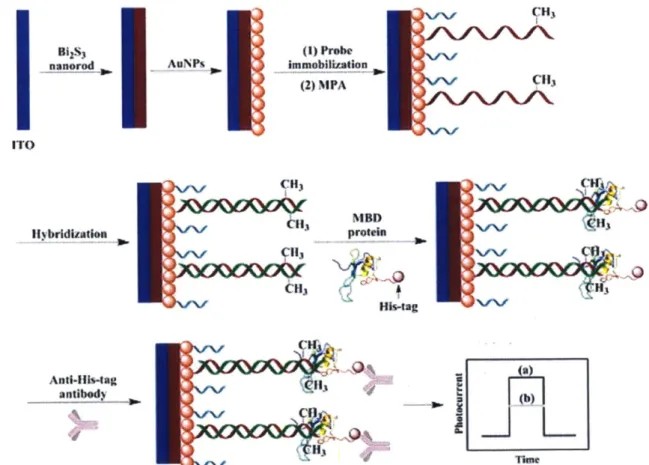
simultaneous analysis of other chromatin modifications as well as selection and recovery of single DNA molecules.41 Other than fluorescence, methods have also been developed that measure the change in photocurrent42 (Figure 1.3) or transport time through a nanopore43 upon MBD binding to methylated DNA. Each of these methods still requires PCR or sequencing, however, for site-specific methylation detection.
Bgs3 nanorod AuNPs I ITO Hybridization CH) 113 Hit3 immobIlization (2) MPA q13 MBD Hb~ag Anti-His-tag antibody 3
Figure 1.3 A schematic showing a method of sequence-specific methylation detection based on the
change in photoelectrochemical response upon MBD and anti-His-tag antibody binding." (Reproduced from Ref. 42 with permission from Elsevier.)
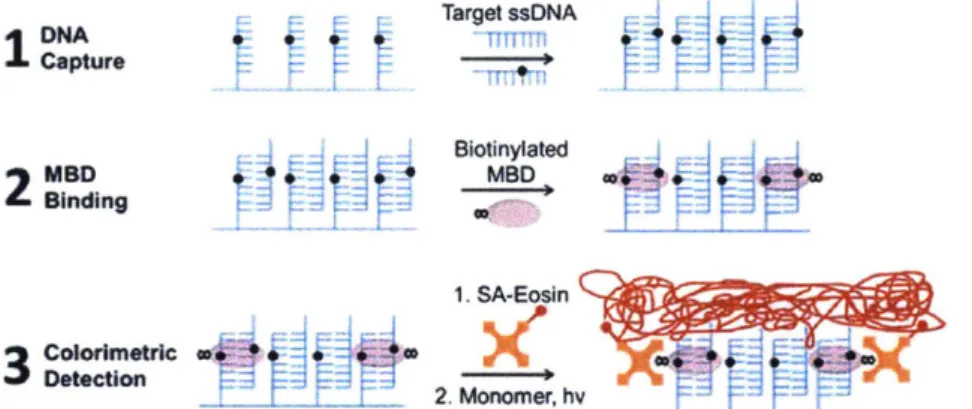
In order to provide sequence-specific DNA methylation analysis, others have proposed capturing target DNA from a sample via direct hybridization to probe ssDNA on a biochip and
detecting methylated CpG sites with MBD proteins. MBD binding is subsequently detected using either surface plasmon resonance (SPR)44 or radical photopolymerization4s (Figure 1.4).
SPR is not ideally suited for clinical analysis because it requires instrumentation not typical for
most pathology labs. Radical polymerization has the advantage of exponential signal
amplification, fast reaction times, and visible readout.46
However, the sample volume and required DNA concentrations necessitate more than 109 copies of each sequence to be interrogated, which equate to gram quantities of tissue to be analyzed. If further work is completed to improve the sensitivity of this and similar methods, they could become valuable
tools in the cancer clinic. One possible route for decreasing the limit of detection is by
incorporating nanoscale features for more efficient DNA capture and MBD binding. Studies have shown that nanoscale curvature can greatly increase the achievable density of DNA probes on a surface4748
while also increasing the hybridization efficiency of target DNA to the immobilized probes.49 This suggests that such features could be used to decrease both the DNA concentration and sample volume required.
Target ssNA Capture Blotinylated
2
MSD~ MBD 1. SA-Eosin r 2. Monomer, hFigure 1.4 A diagram showing sequence-specific methylation detection by capture of the target
sequence on a biochip, MBD binding, and radical photopolymerization.4 5 (Reproduced from Ref. 45 with permission from the Royal Society of Chemistry.)
1.4.4 Methylation Assessment in Fixed Cells
Almost all methods of methylation analysis require purified DNA as a starting point. One
exception to this is a method developed by Li et al.50 called methylation-specific fluorescence in
situ hybridization (meFISH). Shown in Figure 1.5, meFISH is based on standard fluorescence in situ hybridization procedures where a fluorescently labelled DNA probe is hybridized to
genomic DNA within fixed cells in order to detect a sequence of interest. To extend the assay to detect the methylation status of this sequence, the authors used bipyridine-modified DNA probes and osmium oxidation chemistry to crosslink the probes to target DNA containing 5mC. This allowed the researchers to perform a denaturation step that removed all of the DNA probes that were not cross-linked to the genomic DNA so only the methylated sequences
remained labelled. This method not only eliminates both bisulfite treatment and PCR but also the need for DNA purification. However, one major disadvantage is the toxicity of osmium tetroxide. In addition, meFISH has currently only been proven for detecting methylation in satellite repeat sequences where many identical bipyridine-modified probes can bind to amplify the fluorescent signal. Therefore, it is uncertain how feasible this method is for other clinically
relevant sequences such as CpG islands in gene promoters.
Labeled ICON probe --4 -- *
1) In situ hybridization 2) Wash
TTF~Th~1TF~Tfl~
omr~Tn~rn+rrr*
SmC4,
Chromosome cell Tissue sectionTrr1rrr.*
3) Observation of FISH signal 4) Cros.4lnking with osmnm
Tr
&VI*CT
T
5) Denaturation
~rrrfr
4C
&uC
4,
W
8) Observation of MeFISH signalFigure 1.5 A diagram of the MeFISH protocol. After a standard fluorescence in situ hybridization procedure is performed, osmium tetroxide is used to crosslink the bipyridine-modified probe to a methylated CpG. Without a methylated cytosine present, the crosslinking does not occur and the probe is removed upon DNA denaturation.50 (Reproduced from Ref. 50 with permission from Oxford University Press.)
1.5 Clinical Outlook
In order for DNA methylation biomarkers to become part of standard cancer assessment, techniques must be suited to clinical requirements. Clinical sample types range from sputum and stool for early detection of cancers to needle biopsies and surgical pathology specimens.
Each sample has unique characteristics in terms of the amount of DNA available and the
processing required. While the exact sample size depends on many variables including the type
18
and location of the tumor, for a fine needle aspiration biopsy, we can estimate that the number of cells available for molecular testing after preparing the standard smears for morphological studies is on the order of 106 cells.51 While such a sample contains pig levels of DNA, purification procedures result in losses and the sample must be shared among all additional tests that are performed. Often, several molecular assays may be integrated into the diagnostic workup; devoting the entire biopsy sample to one DNA test is infeasible. More sensitive techniques for
DNA methylation detection stand to greatly improve clinical utility of this class of biomarkers.
The ideal DNA methylation analysis technology appears to be one that provides reliable results from very little tissue or body fluid, offers multiplexed analysis, integrates seamlessly into existing pathology practice, and is cost-effective without requiring bisulfite conversion of DNA. Recent advances are moving closer to satisfying all of these criteria. The Cologuard* test, which uses QuARTS, requires bisulfite treatment but addresses this shortcoming by requiring samples be sent to a central test site where experts perform the bisulfite conversion and qPCR-based assay. This strategy raises a particularly salient question, "is outsourced bisulfite-conversion the answer?" In the short term, the answer may be yes. However, it remains an open question whether this operating model will be sustainable from a cost perspective particularly as the number of DNA methylation based assays increases. While many of them are not yet sensitive enough for clinical use, affinity agent-based methods offer the great advantage of simplifying the required detection steps and eliminating the harsh conditions that damage DNA. Recent assay improvements in this area are encouraging. Improving the sensitivity through the use of nanotechnology, new signal amplification techniques, and other assay modifications is an important area of continuing research. In order for DNA methylation to advance in the clinic, these new methods will need to develop simultaneously with the on-going validation of panels of biomarkers to inform clinical diagnosis and management.
1.6 Thesis Organization
This thesis details the engineering of MBD proteins and an investigation into their use in methylation detection assays. Chapter 1 discusses the background related to CpG methylation and its use as a biomarker and highlights some of the existing methods for detecting DNA methylation, along with their advantages and disadvantages. Chapters 2 and 3 are focused on assessing the affinity of MBDs using yeast surface display for equilibrium binding titrations and engineering MBDs with improved binding affinity. Chapter 2 focuses on improving the affinity of MBD binding to symmetrically methylated CpG dinucleotides while Chapter 3 focuses on engineering MBDs with a new capability of binding hemi-methylated CpG dinucleotides. Chapters 4 and 5 discuss using these engineered MBDs in sequence-specific, hybridization-based methylation assays, with Chapter 4 focusing on the experimental investigation of parameters key to improving binding efficiency and Chapter 5 focusing on mathematical
modeling of the thermodynamics and kinetics of MBD-DNA binding reactions. In Chapter 6, I summarize the key achievements of this thesis and discuss directions for future work.
Chapter 2
Characterization and directed evolution of a methyl-binding
domain protein for high-sensitivity DNA methylation analysis
Abstract
Methyl-binding domain (MBD) family proteins specifically bind double-stranded, methylated
DNA, which makes them useful for DNA methylation analysis. We displayed three of the core
members, MBD1, MBD2 and MBD4, on the surface of Saccharomyces cerevisiae cells. Using the yeast display platform, we determined the equilibrium dissociation constant of human MBD2 (hMBD2) to be 5.9 1.3 nM for binding to singly symmetrically methylated DNA. The measured
affinity for DNA with two methylated sites varied with the distance between the sites. We further used the yeast display platform to evolve the hMBD2 protein for improved binding affinity. Affecting five amino acid substitutions doubled the affinity of the wild-type protein to
3.1 1.0 nM. The most prevalent of these mutations, K161R, occurs away from the
DNA-binding site and bridges the N- and C-termini of the protein by forming a new hydrogen bond. The F208Y and L170R mutations added new non-covalent interactions with the bound DNA strand. We finally concatenated the high-affinity MBD variant and expressed it in Escherichia
coli as a green fluorescent protein fusion. Concatenating the protein from 1x to 3x improved binding 6-fold for an interfacial binding application.
2.1 Introduction
The structure of chromatin plays a significant role in gene expression and development for eukaryotic organisms.5 2 Methylation at the 5 position of the cytosine base, when followed by guanine (CpG) in the promoter region of a protein-coding gene, is an epigenetic modification that has been shown to be involved in DNA condensation and transcriptional inactivation.s3 Aberrant DNA methylation patterns have been implicated in the development of human diseases such as cancer.54 Medical research has connected promoter methylation levels for certain genes to therapeutic response in patients. For example, glioma patients with a
methylated promoter for the 06-methylguanine-DNA methyltransferase (MGMT) gene exhibit particular sensitivity to alkylating agent chemotherapeutics,55 and breast cancer patients with methylation-dependent silencing of the breast cancer 1, early onset (BRCA1) gene have been shown to have tumors sensitive to cisplatin.56 Additionally, physicians can test for epigenetic silencing of the DNA mismatch repair gene MutL homolog 1 (MLH1) for its prognostic value for
patients being treated with colon cancer.5", Hypermethylation at glutathione S-transferase pi 1
(GSTP1) has also shown promise as a biomarker for diagnosing prostate cancer. 7 Because promoter methylation has been shown to have predictive, prognostic and diagnostic value, there has been great interest in developing methods for DNA methylation detection with
increased sensitivity, specificity, and resolution to increase clinical value' and also for discovery purposes to generate reference methylome data.5 8
State of the art methods for DNA methylation detection (whole-genome bisulfite sequencing, reduced representation bisulfite sequencing, CpG-specific arrays and methylation-specific PCR) generally rely on sodium bisulfite conversion of unmethylated cytosine bases to uracil.1
Chemical conversion, however, can degrade more than 90% of the sample DNA,19 and protocols must be assiduously optimized to minimize incomplete deamination of unmethylated cytosine bases and inappropriate conversion of methylated ones to thymine.3 Such errors lead to inaccurate results. Alternatively, immunoprecipitation (IP)-based methods such as MeDIP-seq and MBD-seq have been developed. These methods tend to require larger sample inputs5 9 and are not capable of providing single methyl-CpG site resolution without bisulfite conversion.6 0 To avoid bisulfite conversion while still providing improved resolution, there have been several methods developed recently that use the very methyl-binding domain (MBD) proteins involved in forming repressive complexes in vivo to transduce DNA methylation into a signal that can be measured directly4",4 4
,45,6 instead of simply providing sample enrichment as is the case with MBD-seq. These MBD proteins specifically recognize symmetrically methylated CpG
dinucleotides in double-stranded DNA,62
-64 and therefore, have the potential to enable high-resolution DNA methylation detection when paired with sequence-specific probe DNA without requiring chemical conversion or sequencing of DNA. Current MBD-based methods require relatively large amounts of DNA44
,4s,61 or are not sequence specific.40'41 Clinical applications
require that both these problems be addressed.' A very high-affinity MBD protein suitable for interfacial use and capable of recognizing a single methylated CpG site will thermodynamically provide a higher fractional coverage of these sites in DNA,65which is particularly important
when the total number of sites may be low. Such a reagent would support ongoing research to make methylation analysis on a single DNA molecule sequence specific. 4 41 66 6 7
Here, we report the display of MBDs from murine MBD1 (mMBD1), human MBD2 (hMBD2), human MBD4 (hMBD4) and human/murine MeCP2 (h/mMeCP2) on the surface
of Saccharomyces cerevisiae as a platform for systematically characterizing intrinsic binding
properties and engineering variants with improved binding affinity to methylated DNA. We chose the highest affinity wild-type MBD protein, hMBD2, as a parent for directed evolution via error-prone polymerase chain reaction (epPCR) and flow cytometry screening. We isolated
MBD2 variants exhibiting improved binding to methylated DNA and constructed a homology model of each variant using the published chicken MBD2 structure68
to elucidate the molecular basis of the observed affinity enhancements. We further concatenated this MBD variant as a green fluorescent protein (GFP) fusion to create the highest affinity reagent reported for DNA
methylation detection and demonstrated its utility in high-performance interfacial binding applications.
2.2 Materials and Methods
2.2.1 Displaying MBD proteins on the surface of
S.
cerevisiae yeast cells
The cDNA encoding the mMBD1 gene (amino acids 1-75) was PCR amplified from the pET-1xMBD (k19) construct62 from Adrian Bird (University of Edinburgh). The forward 5'-GAC AGC TAG CAT GGC TGA GTC CTG G-3' and reverse 5'-GAC AGG ATC CAG CGT AGT CTG GGA C-3'
primer pair was designed to append flanking 5' Nhel and 3' BamHl restriction sites. The PCR reaction contained Ix Phusion HF reaction buffer (New England BioLabs), 10 nmol of each dNTP (New England BioLabs), 25 pmol of each primer (Integrated DNA Technologies), 10 ng pET-1xMBD construct and 1.0 U Phusion DNA polymerase (New England BioLabs) in a final volume of 50 pl. The thermocycling profile was as follows: initial denaturation at 980C for 30 s followed by 30 cycles of denaturation at 980C for 10 s, annealing at 450C for 30 s, extension at 720C for 30
s and a final extension at 720C for 10 min. The PCR product and pCTCON-2 vector were double
digested with Nhel-HF and BamHI-HF restriction enzymes (New England BioLabs), gel purified, and ligated using T4 DNA ligase (New England BioLabs). The pCTCON-2/mMBD1 construct was transformed into electrocompetent NEB 5-alpha Escherichia coli cells (New England BioLabs). The pCTCON-2/mMBD1 construct was also transformed into EBY100 S. cerevisiae yeast cells
using the Frozen-EZ Yeast Transformation I Kit (Zymo Research) and plated onto SDCAA agar plates.
The cDNA encoding the hMBD2 gene (AAs 145-213) was PCR amplified from the pMal-c2X-MBD2 construct6 9 from Indraneel Ghosh (University of Arizona). The forward 5'-TAC AGC TAG
CGA AAG CGG CAA ACG-3' and reverse 5'-GAC AGG ATC CCA TTT TGC CGG TAC GA-3' primer
pair was designed to append flanking 5' Nhel and 3' BamHI restriction sites. The PCR reaction was carried out as described above. The thermocycling profile was as follows: initial
denaturation at 98"C for 30 s followed by 30 cycles of denaturation at 98'C for 10 s, annealing 24
at 600C for 30 s, extension at 720C for 30 s and a final extension at 720C for 10 min. All other
steps were performed as described above.
The S. cerevisiae codon optimized (Gene Art-Life Technologies) cDNA encoding the hMBD4 (AAs 76-148) and h/mMeCP2 (AAs 78-162) gene including flanking 5' Nhel and 3' BamHI
restriction sites plus four nucleotide overhangs were ordered as gBlocks (Integrated DNA
Technologies). These DNA fragments were double digested, ligated into pCTCON-2 and
transformed separately into both NEB 5-alpha E. coli cells (New England BioLabs) and EBY100 S.
cerevisiae yeast cells. All constructs were verified by sequencing.
2.2.2 Characterizing MBD binding to DNA oligonucleotides with varying methylation
patterns
Quantitative equilibrium binding of DNA to yeast displayed MBD proteins was determined using the method described previously.7 0 EBY100 transformed with pCTCON-2/hMBD2 was grown in
SDCAA media overnight at 30C and 250 rpm. After reaching OD600 = 2-5, cultures were
inoculated to OD6oo = 1 in SGCAA and incubated at 20*C and 250 rpm for 40-48 h to induce
surface display fusion expression. Induced EBY100 was resuspended to OD6oo= 1 in PBSA
(1xPBS, 0.1% (w/v) BSA). 500,000 EBY100 cells in PBSA were incubated with pre-hybridized DNA
(synthesized by Integrated DNA Technologies) at concentrations ranging from 0.06 to 100 nM in
volumes of PBSA ranging from 2225 to 200 pl to provide a 10-fold molar excess of DNA relative to the number of surface display fusions assuming 5 x 104 MBD/cell.70 The DNA
oligonucleotides used for characterizing hMBD2 were derived from the human MGMT gene as described previously" and functionalized with biotin on the 5' end of each target strand to
facilitate fluorescence labeling (Figure 2.1). This oligonucleotide was chosen to determine the
binding affinity of MBD proteins to DNA having a single methyl-CpG dinucleotide and enrich for variants with improved monovalent CpG binding without adding confounding avidity effects
from DNA strands with multiple, methylated CpGs. Further, the sequence complementarity of
this oligonucleotide facilitates interrogation of specific CpG loci when hybridized to sample DNA
for analysis. 5' -TTTGCGGTCCGCTGCCCGACCC-3' 3' -AAACGCCAGGCGACGGGCTGGG-BIO-5'
000
omo 03omm
mom
I0
Figure 2.1 Synthetic DNA oligonucleotides derived from the MGMT gene. All oligos have the same
sequence containing three CpG dinucleotides. The schematic shows the location and number of
methylated CpGs for each test oligo. A 5' biotin was appended to one strand to facilitate detection using a streptavidin conjugated fluorophore.
Equilibrium binding was performed at room temperature for 45 min as described previously.70 The binding of methylated DNA to displayed MBD proteins was detected using streptavidin, Alexa Fluor" 647 (Life Technologies), and the fraction of EBY100 that expressed the surface display fusions was identified using the chicken anti-cMyc (Gallus Immunotech)/Alexa
Fluor" 488 goat anti-chicken (Life Technologies) antibody pair. The dissociation constant (KD) for each oligonucleotide was determined from an equilibrium binding titration curve fit obtained after plotting the mean fluorescence of the EBY100 cells displaying MBDs versus each DNA concentration.70 Each reported KD value is the average of three biological replicates performed on separate days following the same protocol.
2.2.3 hMBD2 library creation using epPCR
The GeneMorph I Random Mutagenesis Kit (Agilent) was used to perform epPCR on the hMBD2 gene. To affect 1-3 mutations per MBD2 gene (-5-15 mutations/kb), 250 ng of target
DNA (7.75 ig plasmid construct) was used as the template for the epPCR reaction. The forward 5'-CGA CGA TTG AAG GTA GAT ACC CAT ACG ACG TTC CAG ACT ACG CTC TGC AG-3' and reverse 5'-CAG ATC TCG AGC TAT TAC AAG TCC TCT TCA GAA ATA AGC TTT TGT TC-3' primer pair70 was used to produce a 367 bp product. The PCR reaction contained 1x Mutazyme I1 reaction buffer (Agilent), 40 nmol of each dNTP (New England BioLabs), 125 ng of each primer (Integrated DNA Technologies), 7.75 Ig pCTCON-2/hMBD2 construct and 2.5 U Mutazyme I DNA polymerase (Agilent) in a final volume of 50 pl. The thermocycling profile was as follows: initial denaturation at 95*C for 2 min followed by 30 cycles of denaturation at 950C for 30 s, annealing at 58*C for 30 s, extension at 72*C for 1 min and a final extension at 72*C for 10 min. The epPCR product was
gel purified and amplified using standard Taq-based PCR to provide sufficient DNA material for library creation via transformation and homologous recombination in EBY100 yeast cells.70
2.2.4 Library screening for MBD2 variants with improved binding affinity to
methylated CpGs
The library was screened using a number of EBY100 cells 10-fold greater than the calculated diversity.70 For the first library, this corresponded to 2 x 109 cells for a diversity of 2 x 108. After
the first round of fluorescence-activated cell sorting (FACS), the number of cells screened was 10-fold greater than the number collected from the previous sort. Because the starting
hMBD2 KD was <10 nM, the library was enriched for high-affinity MBD2 variants using a kinetic screen.7' The library was incubated with 100 nM biotinylated omo dsDNA while ensuring a
10-fold molar excess of DNA for 45 min at room temperature in order to saturate surface displayed MBDs with labeled DNA. The cells were then washed, resuspended in PBSA, and incubated with
100 nM unlabeled, competitor omo dsDNA at room temperature to distinguish clones by the
differences in the degree of labeling due to varying dissociation rate constants and, therefore, binding affinities; concurrently, the cMyc epitope tag of each surface display fusion was labeled with chicken anti-cMyc IgY diluted 1:250. The competition time was determined using the
method described previously" and increased in successive rounds in the range of 90-120 min. The EBY100 population was washed and labeled using streptavidin, Alexa Fluor*647 and Alexa
Fluor® 488 goat anti-chicken secondary reagents (both diluted 1:100) on ice for 15 min. The library was washed and resuspended to a density of 107 cells/ml in sterile PBSF for sorting on a
MoFlo XDP (Beckman Coulter). Diagonal sort gates were drawn to specify the fraction of the cells collected. This value was decreased from 5% to 1%, and finally to 0.1-0.2%, over three consecutive rounds of flow cytometry following the method described previously.7 1 Yeast cells were collected in SDCAA media and subsequently propagated at 300C and 250 rpm. A 10-fold
oversampling of the expanded cells was resuspended in SGCAA media for surface display fusion expression and sorting in the next round of screening. After the third round of FACS, the
plasmids encoding the MBD2-derived variants were collected using the ZymoprepTM Yeast Plasmid Miniprep II kit (Zymo Research) and transformed into Mach 1 E. coli cells (Life
Technologies). Individual clones were isolated and the MBD2 gene was sequenced using the forward primer 5'-CCC CTC AAC TAG CAA AGG CAG-3'.
After screening the first library, the plasmids collected from the final sort were subjected to a second round of mutagenesis by epPCR as described above to create another library with a calculated diversity of 1 x 108. This second library was screened using the same protocol above
for the purpose of finding additional mutations giving rise to higher affinity MBD proteins.
2.2.5 Measuring thermal stability of wild-type and evolved MBDs
Induced EBY100 cells were resuspended to OD600 = 0.5 in PBSA, and samples were prepared for
activity-based thermal stability measurements as follows: 100 p1L of the cell suspension was incubated at 70*C for 10 min and then transferred to a tube containing 1 mL of ice-cold PBSA. Previous studies have demonstrated the stability of the yeast display scaffold at this
temperature. Additional samples were prepared but not exposed to 70*C to determine the activity without thermal degradation. Cells expressing wild-type hMBD2 and variant 1/4 were incubated with 50 nM biotinylated DNA in a total volume of 200 pii, and cells expressing hMBD2 variant 2/5 were incubated with 20 nM DNA in a total volume of 550 Ip1L. These concentrations were determined based on the KD of each protein, and the total volume was chosen in order to
make sure the DNA was present in excess. DNA-binding reactions occurred on ice for 1 h. The
DNA and displayed protein were labeled and analyzed with flow cytometry as described above.
As a measure of protein resistance to thermal degradation, the ratio of fluorescence from displaying cells after exposure to 700C to fluorescence from displaying cells with no thermal
degradation was determined.
2.2.6 Bacterial expression of MBD2 variant proteins
The cDNA for MBD2 variant 2/5 was codon optimized for expression in E. coli (Gene Art-Life
Technologies) and used to create an MBD-GFP fusion analogous to that reported previously.4 4 The protein consists of an N-terminal His6-tag followed by the nuclear localization sequence
A Bsal restriction site was included immediately preceding the MBD2 variant 2/5 to facilitate
concatenation. The cDNA encoding the fusion was synthesized as a gBlock with flanking 5' EcoRI and 3' Xhol restriction sites plus four nucleotide overhangs, double digested, ligated into the
pET-30b+ vector, and transformed into Mach 1 E.coli cells (Life Technologies). The miniprepped
plasmid was subsequently transformed into BL21 (DE3) Tuner E.coli cells (Novagen) for expression.
To create the MBD2 variant 2/5 multimer, we designed a second gBlock consisting of the codon optimized cDNA for the MBD followed by the cDNA for a (Gly4-Ser)2 linker with flanking 5' and
3' Bsal restriction sites plus six nucleotide overhangs on each end. Both the pET-30b+/hMBD2
Variant 2/5 plasmid and second gBlock were digested with Bsal (New England Biolabs) and ligated using T4 DNA ligase (New England Biolabs) such that the digested gBlock was in large molar excess. The ligation product was transformed into Mach 1 E. coli cells and plated onto LB
agar plates supplemented with kanamycin. Individual clones were screened for the number of incorporated MBD Variant 2/5 monomer units on the basis of the size of the fragment obtained following double digestion with EcoRi and Xhol. The plasmid encoding the 3xMBD2 Variant
2/5-GFP protein was transformed into BL21 (DE3) Tuner E. coli cells (Novagen) for expression. The
Ix and 3xMBD2 Variant 2/5 proteins were expressed 7
and purified under denaturing conditions with on-column refolding6 2 using the protocols described previously.
2.2.7 Biochip experiments and affinity determination
Clear glass slides coated with an agarose film were prepared74
and printed45 with
pre-hybridized ooo probe/ooo target, omo probe/omo target and omm probe/omm target oligonucleotides at 10 piM concentration in 3xSSC, as described previously. A circular, 9 mm diameter isolator well was cut from Scotch 3M 665 tape and affixed to the biochip to define each test area. Each biochip was then rinsed under a stream of DI water and blown dry using compressed nitrogen gas. Biochips ready for testing were stored in the vacuum desiccator until needed.
NxMBD proteins were diluted in binding buffer (20 mM HEPES, pH 7.9, 3 mM MgC 2, 10% (v/v)
glycerol, 1 mM dithiothreitol, 100 mM KCI, 0.1% (w/v) BSA, 0.01% Tween-20 and 1 pIM ssDNA) and pre-incubated for 10 min at room temperature. Each 40 pl NxMBD dilution was added to a separate test area and incubated for 40-45 min in a humid chamber at ambient temperature
(-20-22*C). Each slide was washed sequentially with 1xPBS/0.1% (v/v) Tween 20, 1xPBS and 18
MO DI water and blown dry using compressed nitrogen gas. The monoclonal mouse HA.11 clone 16B12 antibody (BioLegend) was diluted 1:100 in PBSA, added to each test area, and incubated for 10 min at 4*C in a humid chamber pre-equilibrated to temperature. The slide was washed and dried as described previously. The secondary Alexa Fluoro 647 goat, anti-mouse antibody was diluted 1:100 in PBSA, added to each test area, and incubated for 10 min at 4*C in a humid chamber pre-equilibrated to temperature. The slide was washed and dried as
described previously before it was scanned with a GenePix 4000B fluorescent microarray scanner (Molecular Devices). Each fluorescence image was analyzed using ImageJ (NIH). The
image to include the entire spot area and averaging the constituent pixel intensities. The values for all spots of the same DNA methylation pattern were averaged and plotted versus the
NxMBD concentration in order to fit the data and determine the apparent equilibrium dissociation constant KD,app.
2.3 Results and Discussion
2.3.1 Yeast surface display and characterization of MBD proteins
We cloned the cDNA encoding the MBD domain from mMBD1 (AAs 1-75),62 hMBD2 (AAs 145-213),63,69 hMBD4 (AAs 76-148)7s and h/mMeCP2 (AAs 78-162)76 into the pCTCON-2 yeast surface display vector. Each of these constructs is expressed as a fusion consisting of Aga2p (for yeast cell surface attachment), HA, MBD and c-Myc.70 Display of each MBD was verified by fluorescently labeling the HA and c-Myc epitope tags with Alexa Fluor' 647 and 488,
respectively, and analyzing with flow cytometry (Figure 2.2). All MBD proteins were successfully displayed on S. cerevisiae strain EBY100 except for h/mMeCP2, which exhibited a truncation likely due to either misfolding or proteolytic cleavage prior to surface display.
hMBD4
0 100 1000 10000 1x105
c-Myc Label (Alexa Fluor 488)
(a)
Aga1d Yeast Surface(d)
(b)
mMBD1 0 100 1000 10000 1x105 c-Myc Label (Alexa Fluor 488)(e)
1x105 100001000
100 < 0 IO 1x106 310000 a 1000 J 100 1 0 0 100 1000 10000 1x105c-Myc Label (Alexa Fluor 488)
1x105 10000 1000
100
II 9100001 1000, 100(c)
hMBD20 100
1000
10000 1x105c-Myc Label (Alexa Fluor 488)
(f)
0 100 1000 10000 1x105 c-Myc Label (Alexa Fluor 488) Figure 2.2 Yeast surface display of MBD family proteins. a) Each MBD protein is displayed as a fusion flanked by N- and C-adjacent HA and c-Myc epitope tags, respectively, on the surface of S. cerevisiae. Each fusion is anchored to the cell surface by disulfide bridges between the Agalp and Aga2p proteins. b-e) Flow cytometry dot plot of yeast displaying b) mMBD1, c) hMBD2, d) hMBD4, and e) h/mMeCP2 with their HA and c-Myc epitope tags labeled with Alexa Fluor* 647 and 488 conjugated antibodies, respectively, for detection of expression. The weak signal from h/mMeCP2 c-Myc labelling indicates a truncated surface display fusion. f) Yeast displaying MBD proteins incubated without either primary antibody but both fluorescently labeled secondary antibodies shows that signal is specific to each epitope tag.
We initially screened the three fully displayed MBD family proteins across a range of methylated DNA concentrations to assess relative binding affinities (data not shown).
Subsequently, equilibrium binding titration was used to quantitatively determine the affinity
and selectivity of the methyl-CpG-binding domain of hMBD2, the highest affinity MBD we
displayed. In addition to an anti-c-Myc/Alexa Fluor" 488 antibody pair used to show surface display expression, yeast were equilibrated with biotinylated DNA at various concentrations followed by secondary labeling with streptavidin, Alexa Fluor*647 (Figure 2.3a). The sequence of the DNA oligonucleotides used in this study was derived from the MGMT gene as described
30 h/mMeCP2 1x105 310000 1000 100 Neg. Ctri. Neither * Ab. Both 20Ab. 7
40
previously4 4 and contains three CpG dinucleotides. Each oligonucleotide was synthesized having
one of four different methylation patterns with no, one or two methylated CpGs (Figure 2.1).
(a) Aga2p Aga P Yeast surface (d) 0 0 U-0 Z 1.0 0.9 0.8 0.7 0.6 0.5 0.4 0.3 0.2 0.1 0.0 (b) r 1x105
a:10000'
1000 Ch M 100 0 * Unmethy1ated (ooo) A* One methylated CpG (omo) A Two methylated CpGs (omm) * Two methylated CpGs (mom)
A A 0--- I *U **.* -(c) S1105
a10000
M
~1000
0~1000
CO Wid-Type hMBD2 50 nM ooo DNA 0 100 1000 10000 1x105cMyc Label (Alexa Fluor 488)
e Wild-Type hMSD2 V MBD2 Var 1/4 * MBD2 Var 2/5 - V -1 0.1 1 10 100 1 10 100 DNA Concentration (nM)
Figure 2.3 Detection and quantification of methylated DNA binding to yeast displayed MBD proteins. a)
Yeast displaying MBD proteins were incubated with biotinylated, methylated DNA and a primary anti-c-Myc antibody followed by labeling with streptavidin, Alexa Fluoro 647 and an Alexa Fluor* 488 secondary antibody, respectively. b-c) Flow cytometry dot plot showing b) 50 nM omo DNA and c) 50 nM ooo DNA
binding to type hMBD2. d) Equilibrium binding titration curves for determining the affinity of wild-type hMBD2 binding to DNA with various DNA methylation patterns. The mean fluorescence of the displaying yeast population is normalized and plotted versus DNA concentration. Fitting the data yields the equilibrium dissociation constant (KD) for each oligo. Each reported value (Table 2.1) is the average of three such biological replicates (only one shown). e) Titration curves for wild-type MBD2, Variant 1/4, and Variant 2/5 binding to omo DNA. Leftward shift of the binding curve indicates higher affinity binding.
The equilibrium dissociation constant for each oligo was determined by fitting the normalized mean fluorescence versus DNA concentration data for each of three biological replicates (Figure
Wild-Type hMBD2 50 nM omo DNA
0 100 1000 10000 1x105
cMyc Label (Alexa Fluor 488)