ROYAUME DU MAROC
UNIVERSITE MOHAMMED V DE RABAT FACULTE DE MEDECINE
ET DE PHARMACIE RABAT
Année: 2020 Thèse N°: 132
LA PRISE EN CHARGE DE L’HYPOCALCÉMIE
CHEZ L’ENFANT (à propos dE 11CAs)
THESE
Présentée et soutenue publiquement le : / /2020
PAR
Madame Doaa EL BAZI
Née le 28 Avril 1995 à Jebha
Pour l'Obtention du Diplôme de
Docteur en Médecine
Mots Clés : Hypocalcémie ; Hypoparathyroïdie ; Pseudohypoparathyoïdie ; vitamine D ; Parathormone
Membres du Jury :
Monsieur Abdelali BENTAHILA
Président
Professeur de Pédiatrie
Monsieur Ahmed GAOUZI Rapporteur
Professeur de Pédiatrie
MonsieurThami BENOUACHANE Juge
Professeur de Pédiatrie
Monsieur Hassan AIT OUAMAR
Juge
Professeur de Pédiatrie
A Allah :
L’unique, le tout puissant,
le clément et le miséricordieux,
qui par sa miséricorde a permi
d’achever cette œuvre.
A l’âme de mon grand-père Mohamed EL BAZI
Malheureusement tu nous as quitté trop tôt, mais c’est le destin
qui en a décidé ainsi. Tu étais un homme unique et extraordinaire
et les valeurs de pardon, de tolérance et de persévérance que tu as semé
en nous nous accompagnent et nous accompagneront pour toujours.
La famille El Bazi te dois beaucoup pour tous tes efforts
de la garder unie et solidaire. Et même si tu n’es plus avec nous,
saches que nous t’aimons et que tu seras toujours
vivant dans nos cœurs.
A ma très chère mère Khadija EL BAZI
Tu représentes pour moi le symbole de la bonté par excellence,
la source de tendresse et l’exemple du dévouement
qui n’a pas cessé de m’encourager et de prier pour moi.
Ta prière et ta bénédiction m’ont été d’un grand secours pour mener
à bien mes études. Aucune dédicace ne saurait être assez éloquente
pour exprimer ce que tu mérites pour tous les sacrifices que tu n’as cessé
de me donner depuis ma naissance, durant mon enfance et même à l’âge
adulte. Tu as fait plus qu’une mère puisse faire pour que ces enfants
suivent le bon chemin dans leurs vie et leurs études.
Je te dédié ce travail en témoignage de mon profond amour.
Puisse Dieu, le tout puissant, te préservera et t’accorder santé,
A mon très cher père Khalid EL BAZI
Tu es pour moi l’homme idéal, l’exemple éternel,
pour toutes les peines et les sacrifices que tu as consentis
pour mon éducation et ma formation. Ce travail ne saurait exprimer
mon amour filial, mon respect et ma profonde reconnaissance.
Aucune expression, ni aucune dédicace ne pourraient exprimer
ce que tu représentes dans ma vie, mais j’espère que tu trouveras ici
dans ce modeste travail le fruit
de tant de sacrifices. Que Dieu te protége et t’accorde santé,
longue vie et bonheur.
A mon très cher époux Imad HAMMOUDAN
Ce travail n’aurait pu voir le jour sans ton aide, ta compréhension ,
ton encouragement et ton soutien. Merci d’être toujours à mes côtés,
dans mes moments de détresse par ta présence, par ton amour dévoué
et ta patience , pour donner du goût et du sens à notre vie de famille.
En témoignage de mon amour, de mon admiration
et de ma grande affection , je te prie de trouver dans ce travail
l’expression de mon estime et mon sincère attachement.
Je prie dieu le tout puissant
pour qu’il nous donne bonheur et prospérité.
A mon très cher frère Diyaa
Les mots ne suffisent guère pour exprimer l’attachement,
l’amour et l’affection que je porte pour vous. Merci pour le soutien,
la tendresse, la serviabilité et le dévouement dont tu m’as fait preuve
le long de mes études. Merci mon très cher frère pour ton affection,
pour ta présence physique et morale à chaque fois que j'en avais besoin.
Je te remercie aussi pour tous les moments de rire et de folie.
J’implore DIEU qu’il t’apporte bonheur, réussite, sérénité
et que tes rêves se réalisent.
Je t’aime mon futur Docteur.
A mon très cher frère Ahmad
Pour les meilleurs et les plus agréables moments. Je te remercie pour ton
soutient sans égal et ton affection si sincère. Merci d’être toujours à mes
côtés, par ta présence pour donner du goût et du sens à notre vie de
famille. Je te dédie ce travail, pour tous les moments de joie et de
taquinerie qu’on partage ensemble. Je t’exprime à travers ce travail mes
sentiments de fraternité, d’amour et d’attachement. Je te souhaite une
vie pleine de bonheur, de santé et de prospérité. .
A mes grands-parents,
A tous mes oncles et tantes maternels et paternels,
A tous mes cousins et toute la famille El Bazi, Taha, Amzour,
Lakhchine, Aadroun, Kadiri, Fariss, Dahman
Je vous remercie pour tous les moments de joie et défets
que nous avons partagés. Je vous remercie également
pour votre perpétuel soutien.
Une dédicace spéciale à ma très chère tante Fadoi EL BAZI
et son mari Si Mohamed KADIRI et leurs enfants Yazid et Mamoun
Merci de m’avoir accueilli durant toutes ces années d’études.
Merci pour vous soutien inconditionnel.
A mes beaux-parents :
Vous m’avez accueillis chez vous, vous m’avez soutenus et aidé
par vos prières et vos encouragements, Je ne pourrais jamais exprimer
le respect que j’ai pour vous. Puisse Dieu, le tout puissant vous
préserver du mal, vous combler de santé et de bonheur.
A mon beau-frère Mohamed, ma belle-sœur Amal,
son mari Abdelhak et leurs enfants Zaid, Nada et Jannat:
Merci de m’avoir accueillir parmi vous.
Vous étiez d’une gentillesse et d’une serviabilité remarquables.
Je vous en serai toujours reconnaissante. Puisse ce travail
témoigner de ma profonde affection et de ma sincère estime.
A ma très chère amie Wijdane El Batane :
A ma meilleure amie et ma confidente, celle avec qui j’ai partagé
mes souvenirs, mes joies et mes tristesses. Tu as toujours
été là pour moi peu importe les circonstances
et en ce jour mémorable, j’aimerai te dédier ce travail en témoignage
de l’amour et de la gratitude pour l’épaule inconditionnelle
que tu représentes pour moi.
À ma très chère amie : Niima Chriki
Grâce à toi, j’ai reconnu le vrai sens
de l’amitié. Tu était toujours-là à m’épauler,
à m’encourager et à rendre ma vie épicée de bonheur, de joie et de folie.
Je t’en serai toujours reconnaissante
En témoignage de l’amitié qui nous uni et des souvenirs de tous les
moments que nous avons passé ensemble,
je te dédie ce travail et je te souhaite
une vie pleine de santé de bonheur et de réussite
A ma chère amie Hidaya Mrabet :
Tu t’es toujours préoccupée de moi en m’octroyant
un soutien morale inestimable et apaisé.
Tu m’as constamment encouragé, on s’est partagé pleins
de choses et aujourd’hui
encore j’aimerais partager avec toi un moment très précieux
dans ma vie ,je te dédie ce modeste travail pour exprimer
ma gratitude et mon amour.
A mes chères amies Mariam El Ghaliou, Sara El Ghaffouli,
Zineb El Bougrini, Soumaya El Graini, Maha el Amani,
Oumaima Belakbir
…. :
On dit que les meilleurs amis sont ceux qui te font rire un peu plus fort,
te font sourire un peu plus longtemps, et te font vivre un peu plus
heureux. Merci pour tous les moments de folie, les fous rires, les
aventures et les découvertes qu’on a partagés, merci d’avoir rendu mes
journées plus joyeuses, et d’être à mes côtés à tout moment.
Je vous souhaite tout ce qu’il y a de meilleur
A tous mes maîtres de l’enseignement primaire,
de l’enseignement secondaire, et de l’enseignement supérieur
En témoignage de mon affection et respect.
A toutes les personnes malades et qui souffrent,
Qu’ALLAH vous garde et vous accorde des jours meilleurs.
A toute personne qui a contribué de près
ou de loin à la réalisation de ce travail.
A notre maître et Président de thèse
Monsieur le professeur Abdelali BENTAHILA
Professeur de Pédiatrie
Il nous fait l’honneur et une grande joie d’accepter
la présidence de notre Jury de thèse, nous en sommes très sensibles.
Vos qualités humaines et vos compétences professionnelles
sont pour nous un exemple à suivre.
Nous espérons égaler un jour votre ardeur au travail
et votre sens de l’humain.
Veuillez agréer, l’expression de notre admiration,
cher Maître, ainsi que notre profonde reconnaissance.
A
notre maître et Rapporteur de thèse
Monsieur le professeur GAOUZI Ahmed
Professeur de Pédiatrie
Nous ne saurions exprimer notre gratitude
pour votre acceptation de nous confier ce travail et de nous
orienter à chaque étape de sa réalisation.
Tous nos remerciements à vous pour votre constante disponibilité
malgré vos obligations professionnelles, votre accueil chaleureux,
votre patience et surtout vos conseils précieux.
Votre dévouement au travail, votre rigueur, votre amabilité
et votre compétence imposent le plus grand respect.
C’est avec un immense plaisir que nous exprimons notre reconnaissance
pour tous vos efforts déployés pour la réalisation de ce travail.
A
notre Maître et Juge de thèse
Monsieur le professeur Thami BENOUECHANE
Professeur de Pédiatrie
C’est avec une profonde gratitude et une joie immense
que nous avons reçu votre acceptation
de faire partie de notre jury de thèse.
Votre présence est, pour nous, l’occasion de vous exprimer
notre admiration de votre grande compétence professionnelle
et de votre grande sympathie.
Que ce travail soit l’humble témoignage de notre gratitude
et notre sincère reconnaissance.
A
notre Maître et Juge de thèse
Madame le professeur Hassan AIT OUAMAR
Professeur de Pédiatrie
C’est un grand honneur et privilège pour nous
d’accepter aimablement et sans réserve de juger ce travail.
Nous portons une grande considération tant pour votre extrême
gentillesse que pour vos qualités professionnelles.
À
Notre Maître et Juge de Thèse
Madame le Professeur TELLAL Saida
Professeur de Biochimie
Nous vous remercions pour la spontanéité avec
laquelle vous avez accepté de juger cette thèse.
Vous nous faîtes un très bon exemple à suivre
par vos compétences et vos qualités morales. Nous avons
bénéficié de votre enseignement lors de notre passage
dans votre service et nous admirons en vous vos qualités
humaines et professionnelles.
Nous vous prions de recevoir ici l'expression
de nos respects les plus considérables.
LISTE DES ABREVIATIONS
AADC : Aromatic L-amino acid decarboxylase.AMPc : Adénosine monophosphate cyclique.
BMI : Body Mass Index.
C3G : céphalosporines de 3ème génération
Ca : Calcium.
CGH array : Comparative genomic hybridization array. DFG : Débit de filtration glomérulaire.
DMP1 : Dentin matrix acidicphosphoprotein 1.
DPM : Développement psycho-moteur.
ECG : Électrocardiogramme.
EEG : Électroencéphalogramme.
ENPP : Ectonucleotide pyrophosphatase/phosphodiesterase.
ERK : Extracellular signal regulated kinase.
ETF : Échographie transfontanellaire.
ETT : Échographie Trans-thoracique.
FISH : Hybridation in situ fluorescente.
GCMB : Glial cells missing homolog b.
GH : Growth hormone.
GNAS : Guanine Nucleotide Binding Protein, Alpha Stimulating.
IGF1 : Insuline-like growth factor 1.
IRM : Imagerie par résonnance magnétique.
KCS : Kenney Caffey Syndrom.
OHA : Pstéodystrophiehéréditaired’Albright.
PDE4D : Phosphodiesterase 4D.
PHEX : Phosphate regulating endopeptidase homolog X-linked.
PHP : Pseudohypoparathyoïdie.
PRKAR1A : cAMP-dependent protein kinase type I-alpha regulatory subunit.
PTH : Parathormone.
PXE : Pseudoxanthome élastique.
SDLR : Signes de lutte respiratoire.
sFRP4 : The protein secreted frizzled protein4.
SGK : Serum glucocorticoid regulated kinase.
SNC : Système nerveux central.
TH : Tyrosine hydroxylase.
TPH : Tryptophan hydroxylase.
TRPM6 : Transient Receptor Potential Melastatin-6.
TRPV : Transient receptor potential channel vanilloid.
VDR : Vitamin D receptor.
VEC : Volume extra cellulaire.
LISTE DES FIGURES :
Figure 1: Embryologie des glandes parathyroïdiennes. ... 12 Figure 2: Représentation schématique de la réabsorption du calcium au niveau de la branche
ascendante large de Henlé. ... 27
Figure 3: Cotransporteurs sodium/phosphate impliqués dans la réabsorption tubulaire du
phosphate ... 32
Figure 4:Patiente numéro 2 ... 49 Figure 5:Patiente numéro 3 ... 55 Figure 6: Évolution de la calcémie après traitement. ... 57 Figure 7 a : Brachydactylie intéressant le 4ème doigt b : brachymétacarpie intéressant le 4ème métacarpien ... 63
Figure 8 : TDM cérébrale coupe axiale sans injection : calcifications des noyaux gris
centraux/ calcifications sous corticales des hémisphères cérébraux. ... 63
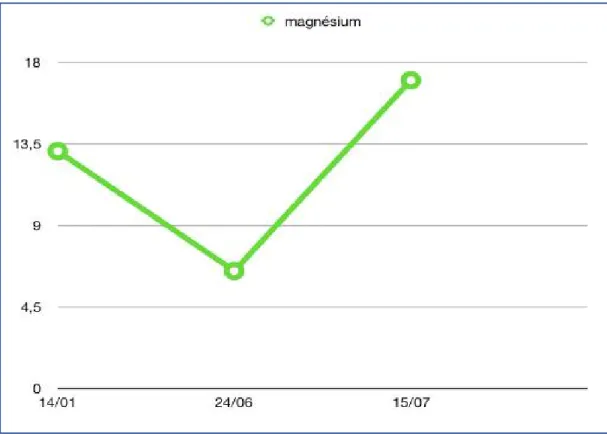
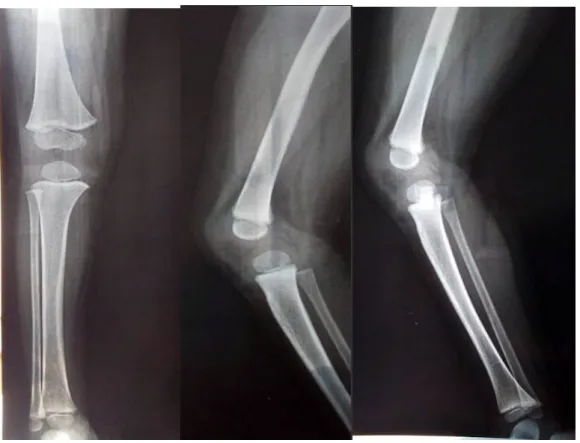
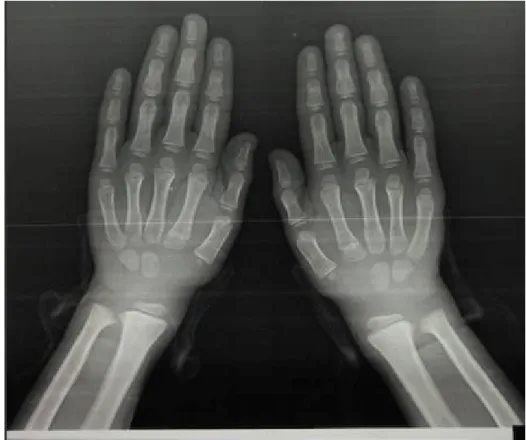
Figure 9: Patient numéro 7 présentant une légère dysmorphie faciale ... 68 Figure 10: Effical, et etalpha ... 71 Figure 11: Cinétique de la calcémie en fonction du temps et après traitement. ... 72 Figure 12: Cinétique de la vitamine D en fonction du temps et après traitement. ... 72 Figure 13: Évolution de la magnésémie en fonction du temps et après traitement... 73 Figure 14: Radiographies de la jambe droite du patient numéro 8. ... 78 Figure 15: Évolution de la calcémie et la vitamine D après traitement. ... 79 Figure 16: Petite taille chez un enfant de notre série ... 80 Figure 17: Radiographie des 2 mains : absence de polydactylie ... 82 Figure 18: Radiographies du bassin face : aspect court des cols fémoraux, et un aspect carré
du bassin. ... 83
Figure 19: ASP face : Aspect en rostre de L4. ... 83 Figure 20: Patient numéro 10 présentant un faciès dysmorphique ... 86 Figure 21: Évolution du poids et la taille en fonction de l’âge ... 88 Figure 22: Variations de la calcémie, la phosphorémie, et la calciurie de 24h en fonction du
Figure 23: Répartition des patients selon l’âge de consultation. ... 97 Figure 24: Répartition des patients selon l’âge de début de la symptomatologie. ... 98 Figure 25: Répartition des patients selon le sexe. ... 99 Figure 26: Répartition des patients selon le motif de consultation. ...100 Figure 27: Répartition des patients en fonction de la consanguinité des parents. ...101 Figure 28: FISH montrant une microdélétion 22q11 ...115 Figure 29: Patient présentant une légère dysmorphie faciale ...116 Figure 30: Représentation schématique de la structure génomique de GATA3 illustrant les
lieux des mutations identifiés dans HDR ...119
Figure 31: Image d’un patient de 26ans avec un KCS type1( microcéphalie, micrognathisme,
hyperpigmentation des contours des yeux avec un nez convexe) ...121
Figure 32: Ptosis bilatéral sévère avec hyper action du muscle frontal. ...123 Figure 33: Rétinographie montrant la fibrose sous-maculaire gauche. ...124 Figure 34: Image objectivant une hypoplasie dentaire chez un enfant ayant le syndrome
APECED ...129
Figure 35: Image d’un patient souffrant du syndrome ayant une candidose buccale ...130 Figure 36 : Image objectivant un vitiligo chez un patient ayant le syndrome APECED ...130 Figure 37 : Syndrome dysmorphique : face ronde, nez bas, et nuque courte. ...132 Figure 38 : Rachitisme carentiel au moment du diagnostic (A) et trois mois après le début du
traitement (B) chez un jeune enfant de 7 ans. ...141
Figure39 : Rachitisme pseudo-carentiel type 2 caractérisé par une alopécie ...145 Figure40: Atteinte osseuse sévère avec nombreuses fractures ...145
LISTE DES TABLEAUX :
Tableau 1: Principales causes génétiques des hypocalcémies ... 8 Tableau 2: Répartition des patients selon l’âge de consultation ... 97 Tableau 3 : Répartition des patients selon l’âge de début : ... 98 Tableau 4: Répartition des patients selon le sexe :... 99 Tableau 5: Répartition des patients en fonction du motif de consultation : ...100 Tableau 6: Répartition des patients en fonction de la consanguinité des parents. ...101 Tableau 7 : Critères diagnostiques du syndrome APECED ...129 Tableau 8: Caractéristiques cliniques, biochimiques et génétiques des troubles
hypoparathyroïdiens et pseudohypoparathyroïdiens : ...134
Tableau 9 : Classification des rachitismes carentiels selon Fraser ...141 Tableau 10: Les caractéristiques biologiques des différents rachitismes ...146 Tableau 11: Principales formes galéniques de calcium injectable ...151 Tableau 12: Formes de sel de calcium les plus utilisées ...152 Tableau 13: Principales formes galéniques du calcium ...152
LISTE DES ANNEXES :
Annexe 1: Valeurs normales phosphocalciques en fonction de l’âge. ...173 Annexe 2 : Bilans à demander en première intention devant une hypocalcémie. ...174 Annexe 3: Conduite étiologique devant l’hypocalcémie. ...176 Annexe 4: Traitement de l’hypocalcémie aigue chez le nouveau-né. ...177 Annexe 5 : Traitement de l’hypocalcémie aigue chez le nourrisson ...178 Annexe 6: Traitement de l’hypocalcémie aigue chez l’enfant. ...179
SOMMAIRE
INTRODUCTION ... 1 HISTORIQUE ... 5 EMBRYOLOGIE ... 10 ANATOMIE DES GLANDES PARATHYROÏDES ... 13
1. SITUATION: ... 14 2. MORPHOLOGIEETSTRUCTURE: ... 14 3. RAPPORTS ... 14 4. VASCULARISATION : ... 15 4.1-VASCULARISATION ARTÉRIELLE : ... 15 4.2-DRAINAGE VEINEUX : ... 15 5.INNERVATION... 15 HISTOLOGIE ... 16 MÉTABOLISME PHOSPHOCALCIQUE... 18 1. LECALCIUM : ... 19 1.1. FORMES : ... 19 1.2. RÉPARTITION : ... 20 1.3. SOURCES ET APPORTS : ... 21 1.4. ABSORPTION INTESTINALE DU CALCIUM : ... 21 1.5. RÉGULATION RÉNALE DE L’HOMÉOSTASIE DU CALCIUM : ... 23 2. LEPHOSPHATE ... 28
2.1RÉPARTITION ET FORMES : ... 28
2.2SOURCES ET APPORTS ... 28
2.3ABSORPTION INTESTINALE DU PHOSPHATE : ... 29
1- AGEDEDÉBUT ETL’ÂGEDECONSULTATION : ... 97 2- SEXE : ... 99 3- MOTIFDECONSULTATION : ...100 4- LESANTÉCÉDENTS : ...101 5- SIGNESCLINIQUES : ...102 6- LESEXAMENSCOMPLÉMENTAIRES : ...102 7- LETRAITEMENT : ...104 8- L’ÉVOLUTIONETSUIVI : ...104 9- DIAGNOSTICÉTIOLOGIQUE : ...105 DISCUSSION ET COMMENTAIRES ...107 1- DIAGNOSTICPOSITIF : ...108 1.1. LA CLINIQUE DE L’HYPOCALCEMIE : ...108 1.2. LA BIOLOGIE DE L’HYPOCALCEMIE :...110 1.3. BILANS MORPHOLOGIQUES DE L’HYPOCALCEMIE : ...112 2- DIAGNOSTICÉTIOLOGIQUE : ...114
2.1HYPOPARATHYROÏDIES GENETIQUES : ...114
2.1.1 Troubles de l’embryogenèse : ...114 2.1.1.1 Syndrome de Di Georges (22q) : ...114 2.1.1.2 L’hypoparathyroïdie isolée (GCMB) : ...118 2.1.1.3 Syndrome HDR : surdité- l’hypoparathyroïdie -anomalies rénales (GATA3) : ...118 2.1.1.4 Syndrome de Kenny-Caffey (SKC) (TBCE, FAM 111A) : ...120 2.1.1.5 Syndrome de Sanjad-Sakati (TBCE) : ...121 2.1.1.6 Syndrome de Kearns-Sayre (mitochondrie) : ...123 2.1.1.7 Syndrome HRD- Hypoparathyroidism, Retardation, Dysmorphism (TCFE) : ...124
2.1.2DÉFAUT DE SYNTHÈSE DE LA PTH : ...125
2.1.2.1Hypocalcémie hypercalciurie dominante autosomique (CaSR, GNAS 11) : ...125 2.1.2.2Hypoparathyroïdie (PTH) : ...126 2.1.3Destruction des glandes parathyroïdiennes par des atteintes auto-immunes : ...128 2.1.3.1Polyendocrinopathie auto-immune de type 1 : APECED (AIRE) ...128 2.1.3.2Anticorps anti CaSR : ...131
2.2 PSEUDOHYPOPARATHYROÏDIES : ...131 2.2.1 PHP type 1A, 1B ou C ...131 2.2.2 Acrodysostose (PRKAR1A) : ...135 2.3 HYPOPARATHYROÏDIES IATROGÈNES : ...135 2.3.1Post-chirurgicales : ...135 2.3.2Post-radiques : ...136 2.3.3Traitement par bisphosphonates : ...136 2.4 HYPOPARATHYROÏDIES AU DECOURS D’AUTRES MALADIES : ...136 2.4.1Insuffisance rénale : ...136 2.4.2Infiltration carcinomateuse : ...137 2.4.3Pancréatite aiguée...137 2.4.4Syndrome de lyse tumorale: ...137
2.5TROUBLES DU METABOLISME DE LA VITAMINE D : ...137
2.5.1Carence d’apports : rachitisme carentiel :...137 2.5.2Rachitisme pseudo-carentiel (VDR, 1 alpha hydroxylase) : (VDR, CYP24A1) : ...142 2.6 HYPOMAGNESEMIES : ...146 3.TRAITEMENTDEL’HYPOCALCÉMIE : ...149
3.1 OBJECTIFS : ...149
3.2 MOYENS :...150 3.2.1Le calcium oral : ...151 3.2.2La vitamine D : ...153 3.2.3Le magnésium : ...155 3.3 INDICATIONS : ...155 3.4 HYPOCALCEMIE AIGUE : ...155 3.4.1 Nouveaux nés : ...155 3.4.2 Nourrisson : ...158 3.4.3 Enfant : ...159
CONCLUSION ...165 RÉSUMÉS ...168 ANNEXES ...172 RÉFÉRENCES ...180
L’hypocalcémie est définit par un taux de calcium plasmatique total inférieur ou égal à 75mg/L ou 1,8mmol/L chez le nouveau-né à terme et chez le nourrisson et l’enfant par un taux égal ou inférieur à 86mg/L ou 2.15mmol/L. [1]
La baisse rapide du taux de calcium moins de 78mg/L ou 1,95mmol/L chez le nourrisson et l’enfant peut avoir des conséquences néfastes sur le SNC, neuromusculaire et la fonction cardiaque. [1]
Un abaissement plus modéré de la calcémie entre 76 et 86 mg/l, soit 1,95 et 2,15 mmol/l ou plus progressif de la calcémie peut à la longue affecter la minéralisation du squelette.[1]
L’hypocalcémie peut apparaître à tout âge suite à de nombreux facteurs environnementaux (augmentation des besoins en calcium liés à la croissance, la carence relative en apports calciques ou en vitamine D) qui peuvent déstabiliser l’équilibre fragile et conduire à une hypocalcémie symptomatique.
Le diagnostic de l’hypocalcémie peut rester longtemps méconnu en raison de la diversité et la non spécificité des symptômes cliniques.
Le tableau clinique est très polymorphe et non spécifique associant des crises convulsives, la tétanie réalisant le spasme carpopédal, les spasmes musculaires avec déformation en main d’accoucheur ( chez le grand enfant). En revanche le laryngospasme est une forme particulière du nourrisson. Les signes cardiaques sont fréquents : tachycardie, voire défaillance cardiaque, l’arrêt cardiaque pouvant surveniret responsable de mort subite.[1] L’importance des signes cliniques dépend largement de la sévérité et la chronicité de l’hypocalcémie. [2]
Le diagnostic repose sur le dosage ( bilan phosphocalcique sanguin et urinaire) de la calcémie, phosphatémie, magnésémie, PAL, PTH, métabolites 1,25 et 25 de la
vitamine D , (voir annexe 1) l’électrocardiogramme qui montre le plus souvent un
allongement de l’espace QT, ou mieux du rapport QT/RR qui s’élève au-dessus de 0,5.[1].
Les autres examens sont demandés en fonction de l’orientation étiologique. Plusieurs étiologies de l’hypocalcémie ont été identifiés, notamment l’hypoparathyroïdie, la Pseudohypoparathyoïdie, les anomalies de la vitamine D, les hypomagnésémies, les causes acquises ( post chirurgicales, irradiation cervicale, post médicamenteuses…) [3]
Dernièrement, de nombreuses études ont montré que la mise en évidence des anomalies moléculaires responsables de certains maladies héréditaires du métabolisme phosphocalcique ont permis de mieux comprendre l’ensemble des acteurs de cette régulation de l’embryogénèse des glandes parathyroïdiennes jusqu’à la valeur de calcémie dosée, d’où l’intérêt de la génétique dans le diagnostic étiologique de l’hypocalcémie. [4]
Devant à une hypocalcémie, le rôle du pédiatre sera de mettre en route immédiatement le traitement pour remonter la calcémie et prévenir les complications cardiaques (troubles du rythme) et de faire le diagnostic étiologique de cette hypocalcémie.
le traitement de l’hypocalcémie est orienté selon l’expression clinique, la gravité et l’étiologie. Le traitement classique repose sur l’association vitamine D dans tous les cas et la supplémentation calcique selon les cas. [3]
Devant les convulsions inexpliqués de l’enfant, il faut toujours vérifier la calcémie.
- Discuter et comparer les résultats avec les données de la littérature.
- Donner les actualités du traitement de l’hypocalcémie.
- Proposer une conduite à tenir pratique en fonction des données de littérature
L’existence des parathyroïdes a été décrite pour la première fois par Richard Owen en 1849 (Professeur au collège royal des chirurgien d’Angleterre) lors de l’autopsie d’un rhinocéros, il les décrit alors comme des formations jaunâtres à l’endroit où les veines émergent du corps thyroïde. [5]
C’est en 1880 que le Dr Ivan Sandström, décrivait la présence constante de glandes parathyroïdes lors de la dissection de cadavres humains. [6][7]
La fonction parathyroïdienne n’a pas été découverte que vers la fin du 19ème
siècle.
A cette époque, on découvrit que les parathyroïdes étaient des entités anatomiques différentes de la thyroïde et que leur absence pouvait provoquer la tétanie.
La structure de la parathormone (PTH) a été établie en 1978 par Keutmann, à l’aide de techniques de séquençage.
Les études fondamentales et le décryptage moléculaire de certaines anomalies héréditaires du métabolisme phosphocalcique, ont permis des progrès énormes dans la compréhension de la physiologie du calcium et des différents acteurs intervenants dans les mécanismes de cette régulation : [4]
- Embryogenèse des glandes parathyroïdiennes.
- Hormones calciotropes et leurs récepteurs, régulation de leur synthèse,
transmission intracellulaire de leur signal hormonal.
- Régulation du turn over des ions calcium aux niveaux intestinal, osseux et
rénal.
Ainsi, de nombreux facteurs de transcription et de gènes intervenant dans la chaîne allant de l’embryogenèse des glandes parathyroïdiennes à la calcémie sont désormais connus, et leur altération peut induire une hyper ou hypocalcémie.
La recherche des anomalies génétiques peut être utile car elle guide la prise en charge immédiate et au long cours, que ce soit sur le plan démarche étiologique, thérapeutique, et évolutif.
Les principales anomalies génétiques actuellement connues responsables d’hypocalcémie sont classées dans le tableau 1.
Tableau 1: Principales causes génétiques des hypocalcémies [8]
Étiologie Mode de
transmission
Localisation
chromosomique Gènes Protéines Mécanisme supposé Caractéristiques
Rachitisme vitaminorésistant type 1[9] AR 11p15.2 1alpha hydroxylase 1alpha hydroxylase
Défaut de synthèse du calcitriol Rachitisme/ostéomalacie, douleurs osseuses… Rachitisme vitaminorésistant type 2[10] AR 12q12-14 VDR Récepteur de la vitamine D
Résistance à la vitamine D par défaut de liaison du calcitriol à son récepteur
Hypocalcémie autosomique dominante[11] AD 3q13-q21 CaSR (mutation activatrice) CaSR (récepteur sensible au calcium)
Gain de fonction du CaSR qui répond à des concentrations de calcium plus basse que la normale
Hypercalciurie inadaptée à l’hypocalcémie, PTH sérique normale, parfois néphrocalcinose et IR
Chondrodysplasie de Biomstrand[12]
AR 3q22-p21.1 PTH-R1 Récepteur de la PTH (type1)
Perte de fonction du récepteur de la PTH
Létale, avance de la maturation osseuse
Hypoparathyroïdie familiale isolée[13]
AD/AR 11q15.3-p15.1 PTH PTH Défaut de synthèse de la PTH Tableau d’hypoparathyroïdie isolée Hypoparathyroïdie isolée[14] AR 6p24.2 GCMB Facteur de transcription GCMB (glial cellmissing B)
Défaut d’embryogénèse des 3ème et 4ème arcs branchiaux
Agénésie des PT Tableau d’hypoparathyroïdie isolée Syndrome HDR (hypoparathyroidis m, deafness, renal defect[15] AR 10p14-15 microdélétion ou mutation ponctuelle GATA 3 Facteur de transcription
Défaut d’embryogénèse des PT, du rein et de l’oreille interne
PT absentes ou hypoplasiques, surdité de perception, dysplasie rénale Syndrome de Di George[16] AD Microdélétion de la région 22q11 Mex40/nex2 TBX1 UDFL1
Défaut de développement de la crête neurale faciale et des 3ème et 4ème arcs branchiaux
Agénésie des PT, et du thymus, cardiopathie, dysmorphie faciale
Syndrome HDR (hypoparathyroidis m, retardation, dysmorphism) Syndrome de Kenny-caffey Syndrome de Sanjad-sakati[17] AR 1q42-43 TCFE Protéine chaperonne
Anomalies de la formation des microtubules intracellulaires impliqués dans les mouvements intracellulaires
Hypoparathyroïdie, RCIU, retard mental et statural, microcéphalie, micropénis, dysmorphie faciale, anomalies osseuses
Pseudohypoparathy oïdie[18]
Empreinte 22q13.3 GNAS Protéine Gs⍺ Atteinte de la transmission postrécepteur de la PTH Hypocalcémie, hypocalciurie, hyperphosphatémie, PTH augmentée Syndrome APECED (auto immune polyendocrinopathy, candidosis, ectodermal dystrophie)[19]
AR 21q22.3 AIRE Protéine AIRE1 :
régule la transcription thymique des auto-anticorps
Atteinte de la tolérance immunitaire Hypoparathyroïdie, candidose cutanéomuqueuse chronique, insuffisance surrénale Syndrome de Kearns-Sayre[20] Maternelle/spo radique ADN mitochondri al
Déficit énergétique cellulaire entrainant un défaut de synthèse de PTH
Hypoparathyroïdie,
hypogonadisme, diabète, atteinte neurologique, oculaire, cardiaque, musculaire, surdité de perception
Les poches entobranchiales sont des invaginations de l’endoderme de revêtement de l’intestin antérieur de l’embryon.
La troisième poche entobrachiale forme un diverticule constitué d’une partie dorsale et qui est à l’origine des glandes parathyroïdes inférieures.
La quatrième proche entobrachiale forme un diverticule présentant une partie dorsale, qui est à l’origine des glandes parathyroïdes supérieures, et une partie ventrale qui donne naissance aux corps ultimo branchiaux.
L’épithélium des ébauches des glandes se différencie en tissu parathyroïdien à la cinquième semaine, les futures glandes perdent leur connexion avec la paroi pharyngienne et migrent en direction caudale et médiane. Les glandes parathyroïdes supérieures et inférieures se mettent sur la face dorsale de la glande thyroïdienne. Les cellules principales et les cellules oxyphiles des parathyroïdes dérivent de l’endoderme. [22][23]
Figure 1: embryologie des glandes parathyroïdiennes.
Représentation schématique des trajets de migration habituels ( flèches A1 et B1) et ectopiques (flèches A2 et B2) 1. Troisième poche endoblastique (P3) ; 2. Pénétration de l’artère thyroïdienne inférieure (ATI) 3. Thymus 4. Quatrième poche endoblastique (P4) 5. Trachée 6.oesophage En bleu : trajet de migration des parathyroïdes inférieures En rouge : trajet de migration des parathyroïdes supérieures.
Anatomie des glandes
parathyroïdes
1. SITUATION :
Les glandes parathyroïdes sont des petites glandes endocrines situées au contact de la face postérieure de la glande thyroïde. Elles sont, en général, au nombre de quatre, une supérieure et une inférieure de chaque côté.
Elles secrètent la parathormone, qui joue un rôle capital dans le métabolisme phosphocalcique.
2. MORPHOLOGIE ET STRUCTURE :
De couleur jaune orangé ou brunâtre, les glandes parathyroïdes mesurent 5 mm de long, 3mm de large et 1mm d’épaisseur . Chaque glande pèse de 35 à 40 milligrammes.
3. RAPPORTS
Les 4 glandes se placent dans une logette fibreuse, pellucide, à l’intérieur de laquelle chaque glande est entourée d’une capsule propre, est légèrement mobile au contact.
Les glandes parathyroïdes sont unies à la face postérieure, et maintenues par un tissu fibreux et des pédicules vasculaires communs aux deux glandes.
Parfois, la glande parathyroïde est enfouie dans un sillon de la glande thyroïde ; parfois, au contraire, elle en est moins solidaire, noyée dans l’atmosphère celluleuse lâche de la gaine thyroïdienne.
La glande parathyroïde supérieure est le plus souvent située en arrière du nerf qui, avant de devenir intra laryngé, est recouvert par le segment moyen du bord postéro-médial de la glande thyroïde.
S’il existe toujours un plan fibreux entre la glande parathyroïde et la glande thyroïde, offrant une possibilité de clivage instrumental entre les deux organes, il n’en reste pas moins que dans la majorité des cas, la mobilisation du lobe thyroïdien entraine les glandes parathyroïdes.
4. VASCULARISATION :
4.1- Vascularisation artérielle :
Chaque parathyroïde est vascularisée par une artère terminale et unique. Les artères thyroïdiennes inférieures vascularisent les parathyroïdes inférieures et 88% des parathyroïdes supérieures. Dans 12% des cas les parathyroïdes supérieures sont vascularisés par l’artère thyroïdienne supérieure ou par l’arcade anastomotique postérieure entre ces 2 artères. Et donc dans la grande majorité des cas, les 4 parathyroïdes sont vascularisées par les seules artères thyroïdiennes inférieures et elles ne reçoivent pas des vaisseaux à partir du parenchyme thyroïdien. Les parathyroïdes médiastinales peuvent être vascularisés par une branche issue de la crosse de l’aorte, du tronc artériel brachiocéphalique ou de l’artère mammaire interne homolatérale. Les parathyroïdes intra thyroïdiennes sont vascularisées par le parenchyme thyroïdien sous adjacent sans pédicule véritablement individualisable.[24][25]
4.2- Drainage veineux :
Il est assuré par un réseau superficiel sous capsulaire qui conflue vers le hile et un réseau profond, de distribution plus variable, non systématisé.[25]
Les parathyroïdes supérieures se drainent vers les veines thyroïdiennes moyennes ou vers le corps de thyroïde.[25]
Les parathyroïdes inférieures se drainent le plus souvent vers les veines thyroïdiennes inferieures.[25]
La cellule principale est de petite taille, de 4 à 8 μm de diamètre ; son noyau est rond. Son cytoplasme est clair, il peut même être vacuolaire à cause des larges plages de glycogène qu'il contient parfois et qui ont été extraites au cours de la préparation.
La cellule principale secrète la parathormone. Le cycle de sécrétion dans cette cellule est particulier, lorsque la cellule est au repos, son cytoplasme est clair et contient de larges plages de glycogène, un appareil de Golgi fortement réduit, ettrès peu de réticulum endoplasmique rugueux.
Quand la cellule est en phase sécrétoire, son cytoplasme est plus foncé, le glycogène ayant disparu, puis le réticulum et l'appareil de Golgi se développant
progressivement. Finalement, des vésicules de condensation et des grains immatures,
au contenu peu dense, se forment dans le cytoplasme.
La cellule oxyphile est plus volumineuse, de 8 à 10 μm de diamètre, son noyau est petit, dense et central, parfois en voie de pycnose. Le cytoplasme est acidophile et granulaire, à cause de ses très nombreuses mitochondries. Le rôle de ces cellules n'est pas connu. Pour certains, elles seraient en voie de dégénérescence ; cependant, leur très grand nombre dans l'adénome parathyroïdien suggère qu'elles ont un rôle dans la synthèse de la parathormone ou dans la régulation de sa sécrétion. [27][28]
Métabolisme
phosphocalcique
1. LE CALCIUM :
Le calcium est l’ion minéral le plus important de l’organisme . il représente à lui seul 50 % des éléments inorganiques de l’organisme. En plus de son rôle fondamental dans la constitution de la structure minérale rigide du squelette, il joue un rôle important dans plusieurs processus biologiques : [29][30]
Excitabilité neuromusculaire. Perméabilité membranaire.
Régulation des activités enzymatiques.
Second messager dans certaines actions hormonales (sous forme ionisée). Coagulation sanguine.
Cofacteur pour un certain nombre d’enzymes.
1.1.Formes :
Le calcium plasmatique est présent sous deux formes :
- 40 à 45 % est lié à des protéines, principalement l’albumine, non ultrafiltrable. - 60% ultrafiltrable :
o 5 à 10 % est lié à des anions.
o environ 50 % est sous forme de calcium ionisé : est le seul physiologiquement actif, efficace et régulé.
Seule la calcémie ionisée est régulée et sa concentration plasmatique est assurée dans des limites très étroites.
Dans les conditions pathologiques, le maintien de la calcémie ionisée peut nécessiter une altération de la balance calcique (c’est-à-dire la différence entre la quantité de calcium qui entre dans le liquide extracellulaire [LEC] et la quantité qui en sort).
La balance calcique est maintenue par trois organes : l’intestin, l’os et le rein. Après un repas, la calcémie augmente transitoirement (c’est pourquoi il faut mesurer la calcémie à jeun). En revanche, à jeun, le maintien de la calcémie dépend seulement de l’équilibre entre la quantité de calcium relarguée par l’os et la quantité de calcium excrétée dans l’urine.
Il existe donc :
- un système régulé, représenté par la calcémie ionisée, et dont la stabilité
dépend de l’équilibre entre les débits d’entrée et de sortie du calcium dans le LEC ;
- un système de stockage représenté par le squelette, où l’organisme va puiser
du calcium quand la calcémie ionisée diminue ;
- un système régulateur, représenté par : PTH, les hormones calciotropes, et
calcitriol (1,25-dihydroxyvitamine D3), qui corrige les variations de la calcémie ionisée détectées par une protéine à sept fragments transmembranaires et le récepteur sensible au calcium (CaSR), présent à la surface des cellules parathyroïdiennes et d’autres tissus dont le rein. [32]
1.2.Répartition :
La quantité totale du calcium est d’environ 2 % du poids total de l’organisme. o Os + dents = 99 % du calcium total.
1.3.Sources et apports :
Les sources alimentaires principales de calcium sont les laitages et les eaux riches en calcium. Les apports nutritionnels conseillés (ANC) sont de l’ordre de 1000 à 1200 mg/j, mais sont variables en fonction de l’âge et du sexe.[33]
- Enfant : 800 mg/j. - Adolescent : 1200 mg/j. - Femme enceinte : 1200 mg/j. - Adulte< 50 ans : 1000 mg/j. - Femme ménopausée : 1200 à 1500 mg/j. - Homme > 65 ans : 1200 mg/j.
Son métabolisme est étroitement lié à celui du phosphore, sous l’influence de 2 facteurs principaux : la parathormone et la vitamine D, et passe par 3 sites principaux : tube digestif, os et rein.
1.4.Absorption intestinale du calcium :
Dans l’alimentation, le calcium est principalement apporté par les laitages et certaines eaux riches en calcium.
Seule une fraction (20 à 60 %) de la quantité de calcium ingérée est absorbée. L’absorption « nette » du calcium correspond à la quantité absorbée moins la quantité sécrétée par les entérocytes vers la lumière intestinale.
Quand la quantité de calcium de la diète augmente, l’absorption dépend de deux processus :
- Un processus passif paracellulaire selon le gradient de concentration et du
gradient électrochimique entre la lumière intestinale et le plasma ;
- Un processus actif transcellulaire médié par la 1,25(OH)2D qui stimule,
dans l’entérocyte, différents gènes dont les produits participent à ce transport actif [34][35] [36]:
1- Une protéine, TRPV6, crée un canal calcium à la bordure en brosse luminale de la cellule intestinale. Le calcium entrant dans la cellule est alors renfermé dans des vésicules qui contiennent une protéine liant le calcium, la calbindine 9K.
2- Les calbindine 9k : protègent la cellule d’un excès de calcium et assurent le transport intracellulaire.
3- Les phosphatases alcalines, la pompe Ca-Mg-ATPase et la pompe Na+/Ca2+ ( de faible affinité mais de forte capacité) : permettent la sortie du calcium vers le plasma.
L’absorption du calcium est stimulée par la 1,25(OH)2D qui active l’entrée du
calcium dans la cellule, augmente l’expression des principales protéines impliquées dans le transport actif de calcium sus cités, ainsi elle favorise à long terme le transfert des cellules des cryptes vers l’extrémité des villosités aboutissant à une modification de la structure des villosités avec différenciation des entérocytes. L’œstradiol augmente aussi l’absorption de calcium par contre les corticostéroïdes l’inhibent.[36]
La capacité de l’intestin à absorber le calcium s’élève quand les apports en calcium ou en phosphate sont inférieurs aux besoins.
Les facteurs alimentaires influencent aussi l’absorption intestinale du calcium [37] :
- Le lactose et les sucres promeuvent l’absorption tout en allongeant le temps
de contact entre le calcium et la paroi intestinale, ainsi l’augmentation du pH intraluminale augmente la solubilité du calcium et donc son absorption.
- L’ingestion de fortes quantités de fibres ( au-dessus de 50g/jr) diminue
l’absorption intestinale du calcium, surtout si ils sont associés à des apports élevées en phytates ( pain complet, riz complet, son, soja), en alginates ( dérivés des algues), ou des apports en uronates>30mmol/jr (légumes et fruits).
L’âge de l’enfant influence considérablement l’absorption de calcium. Elle est plus élevée chez le nouveau-né que chez l’adulte. [37]
1.5.Régulation rénale de l’homéostasie du calcium :
Chaque jour, environ 250 mmol de calcium (il s’agit de la fraction dite « ultrafiltrable » présente dans le plasma) sont filtrés et 98 % du calcium filtré est ensuite réabsorbé le long du tubule rénal pour maintenir la balance calcique.
Environ 70 % du calcium filtré est réabsorbé de façon passive et par voie paracellulaire au niveau du tubule proximal, parallèlement à la réabsorption du sodium.
Grossièrement, 20 % du calcium filtré est réabsorbé au niveau de la branche
Le tubule distal réabsorbe environ 8 % du calcium filtré et est le siège d’une régulation physiologique très fine de l’excrétion urinaire de calcium. La réabsorption du calcium n’y est pas couplée à la réabsorption du sodium et se fait par voie transcellulaire en trois étapes :
- Le calcium entre dans la cellule par l’intermédiaire d’un canal calcium
TRPV5,
- puis est transféré à travers le cytosol jusqu’à la membrane basolatérale par
l’intermédiaire de la calbindin-D28K,
- pour finalement être extrudé hors de la cellule vers l’interstitium par
l’échangeur Na+ –Ca2+ (NCX1) et la Ca2+-ATPase membranaire (PMCA1b).[40]
La régulation de la réabsorption du calcium par le rein est cruciale pour maintenir l’homéostasie calcique. De nombreux facteurs participent à cette régulation, TRPV5 en étant la cible principale :
1/ LaPTH agit sur le néphron à plusieurs niveaux. Elle réduit le débit de
filtration glomérulaire (DFG) et donc la charge filtrée de calcium. Elle stimule la réabsorption du calcium au niveau de la branche ascendante large de Henlé et du tubule distal où elle augmente l’abondance de TRPV5 [22,23]. La PTH-rP a les mêmes effets que la PTH le long du néphron ;
2/ Le calcitriol semble stimuler également la réabsorption tubulaire de calcium.
Il a été montré que, lors d’un déficit en vitamine D, la réabsorption tubulaire de calcium est diminuée, indépendamment du niveau de PTH. De plus, il a été mis en évidence des éléments de réponse à la vitamine D (VDRE) au niveau de la région promotrice du gène de TRPV5, ainsi qu’une augmentation de l’abondance de l’acide ribonucléique messager (ARNm) de TRPV5 et de son expression protéique sous l’effet du calcitriol.[41][42]
3/ Le calcium extracellulaire module la réabsorption du calcium à travers le
Ca-sensingreceptor (CaSR). Ce dernier est exprimé dans presque tous les segments du néphron, mais son expression est la plus intense au pôle basolatéral des cellules de la branche ascendante large de l’anse de Henlé. En cas d’hypercalcémie, le CaSR est stimulé, ce qui diminue la réabsorption de calcium dans ce segment du tubule.[43]
4/ L’augmentation du volume extracellulaire (VEC) réduit la réabsorption
tubulaire proximale de calcium, qui suit celle du NaCl dans cette partie du tubule. La diminution du VEC a l’effet inverse.
5/ La protéine klotho est exprimée principalement dans le tube contourné distal
rénal et, dans une moindre mesure, dans les glandes parathyroïdes et le plexus choroïde cérébral[44][45]. De nombreux arguments semblent impliquer klotho dans la régulation de l’homéostasie phosphocalcique et dans la réabsorption tubulaire du calcium, en particulier. Il a été montré que l’activité b-glucuronidase de la protéine klotho était responsable d’une augmentation de l’expression membranaire de TRPV5. Ainsi, la modification de la glycosylation de TRPV5 par klotho module l’adressage membranaire apical du canal calcium, donc la réabsorption distale de calcium [46] ;
6/ La kallikréine tissulaire est l’enzyme principale permettant la formation de
kinine et est produite dans le néphron distal où elle colocalise avec TRPV5. Les souris invalidées pour la kallikréine développent une hypercalciurie marquée, secondaire à la diminution de la réabsorption rénale de calcium [47]. Il a été montré par la suite que la kallikréine tissulaire stimulait l’activité de TRPV5 par l’intermédiaire de l’activation du récepteur à la bradykinine de type 2. L’activation de ce récepteur, soit directement
7/ L’excrétion urinaire de calcium augmente avec l’acidose et diminue avec
l’alcalose. Le pH extracellulaire module l’activité ainsi que le recrutement à la membrane apicale de TRPV5 selon un mécanisme non connu aujourd’hui : l’alcalinisation du milieu extracellulaire induit un recrutement rapide à la membrane des vésicules contenant TRPV5, alors que l’acidification stimule le retrait de TRPV5 de la membrane plasmique [49] ; TRPV5 est lié à de nombreuses protéines cytoplasmiques, susceptibles de réguler son activité, telles que la protéine 80KH, ou de moduler son adressage à la membrane apicale, comme le complexe protéique S100A10-annexine 2 ou NHERF2, qui est une protéine à domaine PDZ liant les trois derniers acides aminés C-terminaux de TRPV5 [50] ;
8/ Il a été mis en évidence un rôle possible de la WNK (with no lysine [K])
kinase 4 dans l’adressage membranaire de TRPV5. En effet, l’hypertension artérielle familiale en association avec l’hyperkaliémie due à des mutations de la WNK kinase 4 s’accompagne d’une hypercalciurie. De plus, il a été montré in vitro que la WNK kinase 4 stimulait l’expression membranaire de TRPV5 et le transport de calcium dépendant de ce dernier [51];
9/ La magnésémieparticipe à la régulation de la réabsorption rénale du calcium
de façon directe. En effet, il a été montré chez le rat parathyroidïctomisé que la réplétion en magnésium inhibait la réabsorption de calcium au niveau de la branche ascendante large de Henlé [52]. Il se pourrait que cet effet passe par la liaison du magnésium extracellulaire au récepteur sensible au calcium. La magnésémie pourrait aussi réguler la réabsorption rénale du calcium de manière indirecte, par l’intermédiaire de la modulation de la sécrétion de PTH. En effet, les hypomagnésémies sévères conduisent à une inhibition de la sécrétion de PTH[53], donc possiblement a` une diminution de la réabsorption rénale de calcium.
Figure 2: Représentation schématique de la réabsorption du calcium au niveau
2. LE PHOSPHATE
Le phosphate est l’un des minéraux les plus abondants de l’organisme. Il est impliqué dans les échanges énergétiques (adénosine triphosphate [ATP], etc.), certaines activités enzymatiques (phosphatases, phosphorylases), l’équilibre acidobasique, la synthèse des acides nucléiques et le signal intracellulaire (acide adénosine monophosphorique cyclique [AMPc] et guanosine monophosphorique cyclique [GMPc]).
Durant la période de croissance, l’organisme a besoin de quantités importantes de phosphate. On constate d’ailleurs une augmentation de la phosphatémie pendant cette période.
2.1 Répartition et formes :
La quantité totale du phosphore est d’environ 1 % du poids total de l’organisme (500 – 800 g).
- 85% au niveau de l’os sous forme d’hydroxyapatite ou carbonatée lié au
calcium.[54]
- 14% au niveau des tissus mous intra cellulaires.
- 1% au niveau du sang sous forme de :
o Phosphatases organiques. o ATP, phospholipides. o Phosphates inorganiques.
2.2 Sources et apports
Les apports alimentaires sont un élément essentiel dans la régulation du phosphate. Ils varient entre 800 et 2000mg/24h.
L'apport nutritionnel recommandé, à savoir 750 mg /jour de phosphore chez l'adulte en bonne santé, et de 360mg à 800mg/jr chez l’enfant, et il est destiné à maintenir les concentrations sériques de phosphore dans la plage physiologique.[36]
2.3 Absorption intestinale du phosphate :
L'absorption du phosphate a lieu dans tout l'intestin grêle et le côlon. Environ 65% du phosphate ingéré est absorbé.[54]
Il existe deux voies d'absorption intestinale du phosphate :
- Une voie active lors d’un apport faible en phosphate dépendante du
sodium.
- Une voie passive indépendante du sodium quand les apports du phosphate
sont élevés.
Le transport passif du phosphate se fait par transport paracellulaire via les jonctions serrées entre les cellules, tandis que le transport actif s'effectue via le cotransporteur de phosphate de sodium NPT2b et éventuellement les transporteurs de type 3 PiT1 et PiT2.[55]
NPT2b est exprimé dans la bordure de brosse des entérocytes et des appartient à la famille SLC34.[55]
Deux facteurs importants qui régulent l’absorption intestinale du phosphate sont l'apport en phosphate alimentaire et la 1,25-dihydroxyvitamine D3[1,25(OH)2D3].[55] Le transport actif du phosphate est régulé par le 1,25(OH)2D3, induisant l'expression du NPT2b sur la membrane apicale des cellules épithéliales intestinales, et les régimes à faible teneur en phosphate régulent l'expression du NPT2b.
2.4 Régulation rénale du phosphate :
Le rein joue un rôle déterminant dans la régulation de l’homéostasie du phosphate en raison de sa capacité à augmenter ou à diminuer la réabsorption tubulaire en fonction des besoins de l’organisme.
L’excrétion urinaire de phosphate est comprise entre 25 et 33 mmol/j (750 à 1000 mg). Environ 85 % de la réabsorption du phosphate se fait dans le tube
contourné proximal et moins de 10 % est réabsorbé dans les segments tubulaires plus distaux.
L’étape limitante de la réabsorption rénale du phosphate se situe au niveau du pôle apical des cellules tubulaires proximales qui sont le siège d’un cotransport sodium-phosphate.
Une augmentation progressive de la charge de phosphate filtré entraine une augmentation de la réabsorption de phosphate jusqu’à ce qu’un seuil maximal de réabsorption tubulaire du phosphate (TmP) soit atteint. A partir de ce seuil, l’excrétion urinaire du phosphate augmente de façon proportionnelle à la quantité de phosphate filtré. Il existe des variations inter et intra-individuelles du TmP, en partie dues aux variations du DFG. Ainsi, le ratio TmP/DFG indique la capacité maximale de réabsorption tubulaire du phosphate par unité de DFG. Le TmP/DFG est la meilleure estimation de la capacité globale du rein à réabsorber le phosphate et détermine la concentration sérique de phosphate à jeun[56].
Au niveau de la bordure en brosse des cellules tubulaires proximales, quatre cotransporteurs sodium/phosphate ont été identifiés et permettent le transport du phosphate de la lumière tubulaire vers le compartiment intracellulaire (figure 3) : [33]
Le cotransporteur sodium/phosphate de type IIa (NPT2a, SLC34A1) est le
principal déterminant du TmP/DFG. Il a une grande affinité pour le phosphate, et sa haute concentration se trouve dans la membrane de la bordure de la brosse du segment
S1 du tube contourné proximal. Les souris invalidées pour NPT2a présentent une diminution d’environ 80 % du transport tubulaire de phosphate dépendant du sodium, une hypophosphatémie et une hypercalcitriolémie réactionnelle entrainant une hypercalciurie d’origine absorptive et des calcifications rénales[57][58]. L’apport de phosphate dans l’alimentation ou l’invalidation du gène de la 1-a-hydroxylase permettent de réduire, de façon parallèle à la diminution de la calcitriolémie, la calciurie et les calcifications rénales [59],ce qui atteste du rôle fondamental joué par l’hypercalcitriolémie dans le phénotype rénal des souris NPT2a-/-. De la même manière, les patients ayant à l’état hétérozygote des mutations du gène codant pour NPT2a présentent une hypophosphatémie secondaire à une fuite rénale de phosphate et une hypercalciurie pouvant s’accompagner de lithiases rénales[60] ;
Le cotransporteur sodium/phosphate de type IIc (NPT2c, SLC34A3) : il a
une faible affinité pour le phosphate. Ainsi, il est plus abondant dans les reins de jeunes rats que dans les reins de rats adultes, ce qui lui suggère un rôle fondamental pendant la croissance, au cours de laquelle les besoins en phosphate sont accrus [61]. Des mutations à l’état homozygote du gène codant pour NPT2c ont été trouvées chez
des patients atteints d’hypophosphatémie héréditaire avec hypercalciurie
(HHRH)[62][63] . Chez la souris, l’expression de NPT2c est augmentée en réponse à l’invalidation de NPT2a, ce qui ne permet, cependant, pas de restaurer une réabsorption normale de phosphate [64] ;
Les deux transporteurs, NPT2a et NPT2c, sont les principaux transporteurs responsables du contrôle de la réabsorption du phosphate inorganique dans le tubule
Le cotransporteur sodium/phosphate de type 1 (NPT1, SLC17A1) ne
transporte pas spécifiquement le phosphate puisqu’il participe au transport d’autres anions organiques et pourrait également posséder une activité canal chlore[65]. Son rôle exact dans la régulation de l’homéostasie du phosphate reste à déterminer ;
Il a été mis en évidence chez le rat et la souris que Pit2 (SLC20A2), un cotransporteur sodium/phosphate de type III était également localisé à la bordure en brosse des cellules tubulaires proximales et que son expression était régulée par les apports en phosphate.[66].
Figure 3: Cotransporteurs sodium/phosphate impliqués
La régulation de la réabsorption rénale du phosphate est complexe et influencée par différents facteurs, incluant l’apport alimentaire en phosphate, hormones comme FGF23 et PTH, et l’épuisement du volume du liquide extracellulaire.
Les variations de l’apport alimentaire en phosphate modifient le taux de la réabsorption rénale due aux changements de l’expression des transporteurs de phosphate tel que NPT2a, NPT2c, et PiT2, indépendamment de la PTH et 1,25(OH)2D3[67].
La régulation de la réabsorption rénale du phosphate est liée essentiellement à l’adressage à la membrane apicale des cotransporteurs sodium/phosphate, NPT2a
principalement et NPT2c, ou du retrait de ces cotransporteurs de la membrane apicale.
Une fois internalisé , NPT2a n’est pas recyclé , mais il subit une dégradation lysosomale. L’abondance de NPT2a et NPT2c à la membrane apicale des cellules tubulaires proximale est contrôlée par plusieurs facteurs :
1/ Une diète pauvre en phosphate entraîne une augmentation de l’abondance de
NPT2a et du cotransport sodium/phosphate au niveau de la bordure en brosse des cellules tubulaires proximales, mais pas de modification de l’ARNm de NPT2a alors qu’un régime riche en phosphate entraine l’internalisation de la protéine NPT2a, puis sa dégradation lysosomale [68]. Ces modifications de la réabsorption rénale du phosphate en fonction de l’apport en phosphate sont indépendantes de variations de la PTH, de la calcémie ou du VEC ;
2/ La PTH induit l’internalisation puis la dégradation lysosomale de NPT2a,
3/Certaines études ont montré que 1,25(OH)2D3 stimule la réabsorption du
phosphate, en agissant directement sur le tubule proximal à travers l’induction de NPT2a et NPT2c, et semble avoir un rôle facultatif dans l’action de la PTH sur la réabsorption du phosphate [71].
4/Le FGF-23 est une phosphatonine. C’est une glycoprotéine de 21 acides
aminés, secrétée primitivement par les ostéocytes, et dans une moindre mesure par les ostéoblastes, ou les veines sinusoïdes de l’os, les organes endocrines ( parathyroïdes, ovaires, testicules), le cœur, l’hypothalamus, le cortex, l’hippocampe, le putamen, les amygdales, et le noyau ventrolatéral du thalamus, mais l’importance physiologique de FGF-23 produite par ces tissus n’a pas été encore déterminée [72][73].
Dans les conditions physiologiques, FGF-23 est exclusivement produit par les ostéocytes et les ostéoblastes en réponse à une hyperphosphatémie et augmentation de 1,25(OH)2D3 [74].
Le gène codant pour FGF-23 est localisé sur le chromosome 12p12.29.
Le FGF-23 avec son cofacteur Klotho, régule la baisse de l’expression membranaire de NPT2a par une phosphorylation sérique NHERF1par la protéine ERK1/2 et SGK1 dans les tubules proximaux [75].
Par conséquence, la suppression de cotransporteur rénal par FGF-23 entrave la réabsorption rénale du phosphate, ce qui provoque la diminution du taux sérique du phosphate.
La protéine mature FGF-23 (25-251) est sécrétée dans la circulation lors d’un clivage de la séquence signal comprenant 24 acide aminé et O glycosylation par UDP-N-ac2étyl-alpha-d-galactosamine : polypeptide N-acétylgalactosaminyl transférase 3 (GALNT3) [76][77]. L’O glycosylation se produit dans la région 162-228 de FGF-23, et cette modification post traduction la protège contre le clivage par subtilisin-like proprotéine convertase pendant l’utilisation des peptides recombinant in vitro.
Le FGF-23 intact est clivé par une furine-pro protéine convertase en fragments N- et C-terminaux inactifs, et le FGF-23 est ensuite éliminé par le rein. Le peptide signal avec 24 acides aminés se trouve dans la partie N-terminale de la protéine FGF-23, et à côté du peptide signal se trouve la région d'homologie du FGF.
Son peptide C-terminal se lie de façon coordonnée aux récepteurs du FGF (FGFR) dans les tissus ainsi qu'à son co-ligand Klotho, qui existe à la fois sous forme de protéine transmembranaire et sous forme soluble, cette dernière régulant le métabolisme du phosphore par le FGF-23[78][79]
La spécificité des récepteurs du FGF-23 repose sur son interaction avec les récepteurs du FGF (FGFR1, 3 ou 4) et le corécepteur αKlotho, ce qui entraîne l'activation de la voie MAPK (protéine kinase activée par les mitogènes) et du phospho-ERK. Les portions N terminale et C-terminale du FGF-23 sont nécessaires pour cette interaction complète[78].
Bien que la régulation du FGF-23 ait fait l'objet d'un examen approfondi, elle n'a pas encore été entièrement élucidée. Toutefois, le La régulation principale du FGF-23 est basée sur le taux de 1,25(OH)2D3, le phosphate sérique et facteurs locaux[55][80].
Le phosphate sérique et le 1,25(OH)2D3 sont tous des régulateurs positifs du FGF-23 ; lorsque les niveaux de phosphate sérique ou de vitamine D sont élevés, le niveau de FGF-23 s'élève pour augmenter l'excrétion rénale du phosphate et pour diminuer le taux de 1,25(OH)2D3 actif.
En particulier, 1,25(OH)2D3 régule le FGF-23 via le des mécanismes dépendants et indépendants du récepteur de la vitamine D (VDR).


![Figure 3: Cotransporteurs sodium/phosphate impliqués dans la réabsorption tubulaire du phosphate [33]](https://thumb-eu.123doks.com/thumbv2/123doknet/14385321.700302/77.892.85.807.415.863/figure-cotransporteurs-sodium-phosphate-impliqués-réabsorption-tubulaire-phosphate.webp)