Biomedical Data Sharing and Analysis at Scale:
Privacy, Compaction, and Integration
by
Hyunghoon Cho
B.S., Stanford University (2013)
M.S., Stanford University (2013)
Submitted to the Department of Electrical Engineering and Computer Science
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
June 2019
@
Massachusetts Institute of Technology 2019. All rights reserved.
Signature redacted
Department of Electrical Engineering and Computer Science
Certified by.
Signature
May 23, 2019
red acted
Accepted by .... MASSACHUSTS INSTI OF TECHNOLOGYJUN 13 2019
LIBRARIES
Bonnie Berger
Simons Professor of Mathematics
Professor of Electrical Engineering and Computer Science
Thesis Supervisor
Signature redacted
TUTE
Leslie A. Kolodziejski
Professor of Electrical Engineering and Computer Science
Chair, Department Committee on Graduate Students
ARCHIVES
Author
Biomedical Data Sharing and Analysis at Scale:
Privacy, Compaction, and Integration
by
Hyunghoon Cho
Submitted to the Department of Electrical Engineering and Computer Science on May 23, 2019, in partial fulfillment of the
requirements for the degree of Doctor of Philosophy
Abstract
Recent advances in high-throughput experimental technologies have led to the exponential growth of biomedical datasets, including personal genomes, single-cell sequencing experi-ments, and molecular interaction networks. The unprecedented scale, variety, and distributed ownership of emerging biomedical datasets present key computational challenges for sharing and analyzing these data to uncover new scientific insights. This thesis introduces a range of computational methods that overcome these challenges to enable scalable sharing and analysis of massive datasets in a range of biomedical domains. First, we introduce scalable privacy-preserving analysis pipelines built upon modern cryptographic tools to enable large amounts of sensitive biomedical data to be securely pooled from multiple entities for col-laborative science. Second, we introduce efficient computational techniques for analyzing emerging large-scale sequencing datasets of millions of cells that leverage a compact sum-mary of the data to speedup various analysis tasks while maintaining the accuracy of results. Third, we introduce integrative approaches to analyzing a growing variety of molecular in-teraction networks from heterogeneous data sources to facilitate functional characterization of poorly-understood genes. The computational techniques we introduce for scaling essential biomedical analysis tasks to the large volume of data being generated are broadly applicable to other data science domains.
Thesis Supervisor: Bonnie Berger
Title: Simons Professor of Mathematics and Professor of Electrical Engineering and Com-puter Science
Acknowledgments
The completion of this thesis and my PhD studies would not have been possible without the support of my mentors, colleagues, friends, and family both in and out of MIT.
First and foremost, I am deeply thankful to my advisor, Bonnie Berger, for all she has done to propel and guide me through every step of my PhD and personal growth as an academic. Her advice and encouragement gave me the strength to take on challenges that helped push my limits. As I embark upon my own path to mentoring the future generations of researchers, her passion for research, foresight, and unwavering support for students will remain constant sources of inspiration for me.
I am fortunate to have been surrounded by the wonderful people of the Berger lab, both past and present. I thank Sumaiya Nazeen, Deniz Yorokoglu, William Yu, Sean Simmons, Yaron Orenstein, Tristan Bepler, Perry Palmedo, George Tucker, Noah Daniels, Brian Hie, Ben DeMeo, Ibrahim Numanagic, Ariya Shajii, Sarah Nyquist, Max Sherman, Ellen Zhong, Ashwin Narayan, Younhun Kim, and many other members of the lab for stimulating discus-sions about life and research as well as fun activities outside of work, including post-deadline celebrations, Disney movie outings, and conference adventures. I want to specially thank Jian Peng for his mentorship and support and Patrice Macaluso for her help, warm presence, and brownies (which got me through Mondays during the first few years of PhD).
I am also grateful for my friends outside of the lab for their support. I thank Albert Kim, Atalay Ileri, Haoyang Zeng, and Menghan Li for their company and numerous excursions and boardgame nights that helped me keep my sanity. I am also grateful to James Noraky, Orhan Celiker, and Madalina Persu for cherishable memories. I thank my Korean friends Hyodong Lee, Albert Kwon, Kyel Ok, and Taehong Kwon for reminding me of home. I also want to thank my friends and mentors from the music community at MIT (Joonwon Choi, Alex Wang, Carina Belvin, Roger Levy, Katherine Young, Stephanie Doong, Eugene Kim, Jean Rife, and Marcus Thompson) and the MIT Aikido Club (Zhe Lu, Olimpia Ziemniak, Carmen DiMichele, Matthew Tom, Bob Toabe, Matthew Bagedonow, and Mitchell Hansberry) for inspiring me in ways that complemented and enriched my PhD experience and helped me grow as a person. My longtime friends from Stanford, David Wu and Triwit Ariyathugun,
have also provided immense support throughout my PhD, for which I am grateful.
I would not be where I am if it wasn't for my parents, Dongsub Cho and Mija Lee, and my sister Hyejin Cho, who have been sending their love and support (and occasionally food and fashionable clothes) from South Korea ever since I moved to the United States in 2009. I am also grateful for my brother-in-law Changdo Park for his warm support. I dedicate this thesis to my niece Seoyeon Park, who may one day very well become a researcher herself if she wishes so-perhaps even a computational biologist!
Lastly, I want to express sincere gratitude for my thesis committee members, Vinod Vaikuntanathan and Devavrat Shah, for their consideration and feedback.
Previous Publications of This Work
Chapter 3 includes results from the following publications:* Hyunghoon Cho, David J. Wu, Bonnie Berger, "Secure Genome-wide Association Anal-ysis Using Multiparty Computation," Nature Biotechnology 36(6), 2018.
* Brian Hie*, Hyunghoon Cho*, Bonnie Berger, "Realizing Private and Practical Phar-macological Collaboration," Science, 362(6412), 2018.
Chapter 4 includes results from the following publications:
" Hyunghoon Cho, Bonnie Berger, Jian Peng, "Generalizable and Scalable Visualization of Single Cell Data Using Neural Networks," Cell Systems 7(2), 2018. Also appeared in RECOMB 2018.
" Brian Hie*, Hyunghoon Cho*, Benjamin DeMeo, Bryan Bryson, Bonnie Berger, "Ge-ometric Sketching of Single-Cell Data Preserves Transcriptional Structure," Cell
Sys-tems, 2019. Also appeared in RECOMB 2019.
Chapter 5 includes results from the following publications:
" Hyunghoon Cho, Bonnie Berger, Jian Peng, "Compact Integration of Multi-Network Topology for Functional Analysis of Genes," Cell Systems 3(6), 2016.
* Sheng Wang*, Hyunghoon Cho*, ChengXiang Zhai, Bonnie Berger, Jian Peng, "Ex-ploiting Ontology Graph for Predicting Sparsely Annotated Gene Function,"
Bioinfor-matics 31(12), 2015. Also appeared in ISMB/ECCB 2015.
" Hyunghoon Cho, Bonnie Berger, and Jian Peng, "Diffusion Component Analysis: Un-raveling Functional Topology in Biological Networks," RECOMB 2015.
Contents
1 Introduction 19
1.1 The Era of Big Biomedical Data. . . . . 19
1.2 Key Challenges and Motivation . . . . 21
1.3 Contributions of This Research . . . . 22
1.4 Roadmap of Thesis . . . . 24
2 Background 27 2.1 Genome-wide Association Studies . . . . 27
2.2 Drug-Target Interaction Prediction . . . . 29
2.3 Single-Cell Transcriptomics . . . . 31
2.4 Network Biology . . . . 33
3 Privacy-Preserving Biomedicine at Scale 35 3.1 O verview . . . . 35
3.2 Secure Genome-wide Association Studies . . . . 36
3.2.1 Introduction . . . . 36
3.2.2 Our Approach: Secure GWAS . . . . 38
3.2.3 Methods . . . . 41
3.2.4 R esults . . . . 45
3.2.5 Discussion . . . . 50
3.3 Secure Pharamacological Collaboration . . . . 52
3.3.1 Introduction . . . . 52
. . . . 5 5
3.3.4 R esults . . . . 63
3.3.5 D iscussion . . . . 67
3.4 Our Secure Computation Framework Details . . . . 68
3.4.1 Protocol Setup . . . . 68
3.4.2 N otation . . . . 70
3.4.3 Secret Sharing . . . . 70
3.4.4 Computing on Secret-Shared Data . . . . 72
3.4.5 Generalizing Beaver Multiplication Triples . . . . 74
3.4.6 Protocol Building Blocks . . . . 83
3.4.7 Secure Computation with Fixed-Point Numbers . . . . 85
3.4.8 Choice of Base Primes . . . . 98
3.4.9 Further Optimization with Shared Random Streams . . . . 100
3.4.10 Secure Linear Algebra . . . 102
3.4.11 Secure GWAS Protocol . . . 106
3.4.12 Secure DTI Protocol . . . 121
3.5 Caveats and Alternative Approaches . . . . 124
3.5.1 Towards Logistic Regression Analysis for GWAS . . . . 124
3.5.2 Relaxing the No-Collusion Assumption in the Online Phase . . . 128
3.5.3 Handling Active Adversaries in the Online Phase . . . . 128
3.5.4 Other Cryptographic Frameworks . . . . 132
4 Single-Cell Data Analysis at Scale 135 4.1 O verview . . . 135
4.2 Generalizable Data Visualization of Single Cells . . . 137
4.2.1 Introduction . . . 137
4.2.2 Our Approach: net-SNE . . . 138
4.2.3 M ethods . . . 139
4.2.4 R esults . . . 145
4.2.5 D iscussion . . . 153
10 3.3.3 Methods . . .
4.3 Sketch-based Acceleration of Large-Scale Single-Cell 4.3.1 Introduction . . . . 4.3.2 Our Approach: GeoSketch . . . . 4.3.3 Methods . . . . 4.3.4 R esults . . . . 4.3.5 Discussion . . . .
5 Biological Network Analysis at Scale
5.1 O verview . . . . 5.2 Compact Integration of Heterogeneous Interaction
5.2.1 Introduction . . . . 5.2.2 Our Approach: Mashup 5.2.3 Methods . . . . 5.2.4 Results . . . . 5.2.5 Discussion . . . . 5.3 Hierarchical Classification of Gene
5.3.1 Introduction . . . . 5.3.2 Our Approach: clusDCA . 5.3.3 Methods . . . . 5.3.4 Results . . . . 5.3.5 Discussion . . . .
6 Conclusion and Future Work
A Supplementary Tables B Supplementary Figures Networks . . . . . . . . . . . . . . . . . . . .
Function via Compact
. . . . . . . . . . . . Representations 11 Analysis 154 154 157 157 169 181 185 185 187 187 187 190 200 211 212 212 215 217 222 226 229 231 249 .
List of Figures
1-1 Overview of this thesis . . . .
Illustration of GWAS . . . . Illustration of drug-target interaction (DTI) prediction Illustration of single-cell RNA sequencing . . . . Visualization of the yeast interactome . . . .
3-1 Overview of our secure GWAS pipeline . . . . 3-2 Our secure GWAS protocol obtains accurate results . . 3-3 Our secure GWAS protocol achieves practical runtimes,
bility metrics follow a linear trend . . . . 3-4 Secure pipeline for pharmacological collaboration . . . 3-5 Prediction of drug-target interactions . . . .
and all of . . . . 39 . . . . 48 our scala-. scala-. scala-. scala-. scala-. scala-. 49 . . . . 54 . . . . 64 3-6 Call graph of subroutines that utilize the auxiliary finite field . . . .
4-1 The increasing scale and redundancy of single-cell RNA-seq datasets . . . . . 4-2 net-SNE recapitulates t-SNE mapping on 13 benchmark datasets with known
subtypes ... ...
4-3 net-SNE generalizes to unseen cells . . . . 4-4 net-SNE enables fast visualization of mega-scale datasets . . . . 4-5 Illustration of Geometric Sketching . . . . 4-6 Geometric sketching yields more even coverage of the transcriptomic space 4-7 Geometric sketches contain more balanced summaries of the transcriptional
landscape ... ... 2-1 2-2 2-3 2-4 23 28 30 32 34 99 147 148 150 152 156 170 172
4-8 Geometric sketches preserve rare cell types in the subsampled data . . . 174
4-9 Geometric sketching is consistently effective at distinguishing biological cell types via clustering . . . 176
4-10 Novel subpopulation of inflammatory macrophages identified using Geometric Sketching . . . 178
4-11 Geometric sketching efficiently scales to large single-cell datasets . . . 180
4-12 Geometric sketching accelerates single-cell data integration tools . . . 182
5-1 Overview of M ashup . . . 189
5-2 Mashup improves gene function prediction performance in human and yeast 202 5-3 Integrating multiple networks outperforms individual networks in gene func-tion predicfunc-tion . . . 203
5-4 Mashup improves network-based gene ontology reconstruction . . . 206
5-5 Mashup improves genetic interaction prediction and drug efficacy prediction 209 5-6 A breakdown of GO labels by the number of annotated genes in human and yeast . . . 2 13 5-7 Overview of clusDCA . . . 216
5-8 Function prediction performance of clusDCA . . . 224
5-9 clusDCA accurately predicts genes for previously unseen GO labels in yeast 226 B-1 Secure calculation of GWAS odds ratios . . . . 250
B-2 Performance evaluation of Secure DTI on DrugBank dataset . . . 251
B-3 Performance evaluation of Secure DTI on STITCH dataset . . . 252
B-4 Secure DTI performance increases with dataset size . . . 253
B-5 Experimental validation of our DTI predictions . . . 254
B-6 net-SNE visualizations of benchmark datasets and model architecture . . . . 255
B-7 Accuracy of hierarchical clustering of net-SNE embeddings . . . 256
B-8 Robustness of net-SNE performance to neural network size . . . 257
B-9 Generalization performance of net-SNE . . . 258
B-10 Visualizations of different sketches of large-scale scRNA-seq datasets . . . 259
B-11 Geometric sketching better reflects differences in cluster volume instead of
density . . . .. .. . . . ... . . . . 260
B-12 Rarest cell types are more represented within a geometric sketch . . . . 261
B-13 Unbalanced measurement of clustering recapitulation of biological cell types 262 B-14 Unique gene heatmap of umbilical cord cells . . . . 263
B-15 Integration quality of methods with and without geometric sketch-based ac-celeration . . . . 264
B-16 Near monotonicity of covering boxes with box length . . . . 265
B-17 SVD runtime versus dataset size . . . . 266
B-18 Partial Hausdorff distance at different parameter cutoffs . . . . 267
B-19 Low fractal dimension of single-cell data . . . . 268
B-20 Comparison with clustering-based sampling methods . . . . 269
B-21 Mashup's function prediction outperforms other data integration approaches 270 B-22 Mashup outperforms other methods on human GO molecular function (MF) and cellular component (CC) datasets . . . 271
B-23 Mashup demonstrates superior precision-recall curves over other methods for function prediction in human . . . 272
B-24 Robustness of Mashup to restart probability and the number of dimensions for function prediction . . . 273
B-25 Robustness of Mashup to number of dimensions for genetic interaction prediction274 B-26 Mashup outperforms a modified DSD method that approximates Mashup without dimensionality reduction . . . 275
B-27 Robustness of Mashup to random perturbation of edges in the network data 276 B-28 Mashup outperforms CliXO at ontology reconstruction on YeastNet dataset on all ontologies but biological process (BP) . . . . 277
B-29 Comparison of yeast genetic interaction prediction performance on Costanzo et al. data ... ... 278
List of Tables
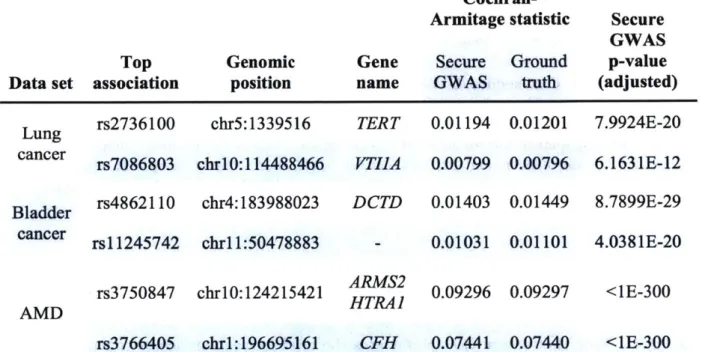
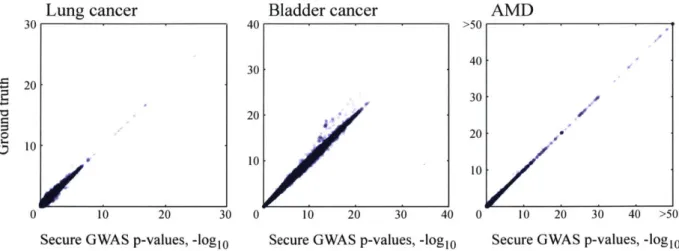
3.1 Our secure GWAS protocol identifies SNPs with significant disease associa-tions while protecting privacy . . . . 47 3.2 Predicted out-of-dataset drug-target interactions . . . . 66 3.3 Examples of tuple notation for partial data visibility . . . . 70 3.4 Comparison of Beaver multiplication triples and our generalized method for
arithm etic circuit evaluation . . . . 80 3.5 Comparison of Beaver multiplication triples and our generalized method for
m atrix m ultiplication . . . . 81 3.6 Comparison of Beaver multiplication triples and our generalized method for
pow er iteration . . . . 82 3.7 Comparison of Beaver multiplication triples and our generalized method for
exponentiation . . . . 83 3.8 Communication complexity of our improved method combining our Beaver
partitioning method and shared random streams . . . 101
5.1 Number of gene ontology (GO) terms in different species and sparsity levels . 220
A. 1 Top 20 associations obtained by our secure GWAS protocol on lung cancer dataset . . . .. . . .. . . . 232 A.2 Top 20 associations obtained by our secure GWAS protocol on bladder cancer
dataset . . . ... ... .. .. .. . . . .. . . .. . . . ... 233 A.3 Top 20 associations obtained by our secure GWAS protocol on AMD dataset 234 A.4 Runtime and communication of Secure DTI . . . 235 A.5 Novel DTI predictions by Secure DTI-A based on the STITCH database . . 236
A.6 Novel DTI predictions by Secure DTI-B based on the STITCH database . 237
A.7 Input feature importance analysis of the top predictions of Secure-DTI-A . 240
A.8 Benchmark single-cell RNA-seq datasets for the evaluation of net-SNE . . . 241
A.9 Statistics for 293/Jurkat mixture data . . . 242
A.10 Statistics for PBM C data . . . 243
A.11 Statistics for adult mouse brain data . . . . 244
A.12 Statistics for developing and adolescent mouse CNS data . . . 245 A.13 Number of cells from each cell type in subsamples visualized in Figure 4-7. 246 A.14 Number of cells from each cell type in subsamples visualized in Figure 4-7. 247 A.15 Drugs with significant efficacy prediction and predicted SDL interactions . 248
Chapter 1
Introduction
1.1
The Era of Big Biomedical Data
Our knowledge of biology and medicine, akin to other scientific domains, are built upon
quantitative observations about the world. In a study of cancer, for example, one may
collect information about genetic mutations that frequently appear in cancer cells, profile the biochemical properties of cancer-associated proteins and their interactions with drug compounds, or record clinical variables across individual patients at hospitals. Analyzing and identifying meaningful patterns in these observations, so-called 'data,' is an essential component of modern biomedicine, emboldened by the belief that the information contained within the data could be distilled into novel biological or medical knowledge that can help save lives.
In the last two decades, advances in experimental technologies have transformed the landscape of biomedical data at a remarkable pace. A prominent example is the dropping cost of sequencing: determining the sequence of a human genome, which was once an inter-national effort-namely, the Human Genome Project [601-that cost billions of dollars and took multiple years, now costs only a thousand dollars and hours of time
1276].
As a result, hundreds of thousands of private individuals are gaining access to their own genome through commercial companies, and more research labs and institutes around the world are estab-lishing their own sequencing facilities, leading to a highly distributed landscape of genetic data.In addition to the enhanced throughput and accessibility of sequencing pipelines, the
biological context from which these measurements are taken are becoming increasingly
fine-grained. Techniques for isolating and tagging individual cells in a sample so they can be separately characterized, as opposed to the traditional approach of collectively measuring a population of cells, gave rise to the field of 'single-cell genomics' [95], which studies the biological processes in a cell and their relations to the genome at a single-cell resolution. Recent developments in this domain enabled millions of cells to be sequenced in a single experiment and allowed multi-faceted characterization of cells that additionally incorporates spatial information, genetic perturbations, or surface interactions. Moreover, academic con-sortia have been formed under the vision of creating a comprehensive reference map of billions of cells across diverse tissues in human body, reflecting the community's enthusiasm for this technology [219].
Recent advances that have pushed the scale of biomedical data is not limited to sequenc-ing. High-throughput screening technologies allowed a comprehensive characterization of molecular interactions in various model organisms such as yeast and helped to launch the field of 'interactomics,' which investigates the properties of genes or proteins in the context of their interaction network as a whole. Though noisy and incomplete, interaction networks in human have also been extensively used to derive biomedical insights, and recent studies have expanded the range of available networks to tissue-specific contexts [106] as well as heterogeneous data sources
188],
including functional interactions and correlated evolution. High-throughput screening platforms have also been an important driving factor in mod-ern drug discovery pipelines by enabling researchers to assess the activity of hundreds of thousands of chemical compounds on biological systems of interest in a matter of days.The emergence of these data collections (or 'datasets') of unprecedented scale-commonly referred to as 'big data' in the broad context including artificial intelligence and customer analytics systems-signals the beginning of an exciting new era for biomedicine, in which the sheer amount, resolution, and variety of emerging datasets enable researchers to an-swer important scientific questions whose anan-swers were previously beyond reach due to the limitations of the data.
1.2
Key Challenges and Motivation
A major consequence of the rapidly growing scale of biomedical data is that data generation is no longer the rate-limiting step for biomedical research. Instead, the main bottleneck is now the ability to share and analyze large amounts of biomedical information. This transition engendered pressing needs in the field for effective computational tools for data sharing and analysis that can help researchers to extract information from increasingly distributed,
large-scale, and multi-modal biomedical datasets. Each of these aspects presents a unique
computational challenge as described below.
First, the scattered generation of biomedical data across many different labs, institutes, and hospitals around the world has made data sharing a necessity in the field. Data sharing allows researchers to acquire sufficient data for gaining fine-grain insights into human biology and its impact on health and disease. The biomedical community has widely recognized the need for data sharing, which has recently led to several national and international collabo-rative research partnerships in various domains [246, 96, 50, 86, 1011. However, the sensitive nature of biomedical information [173] restricts the scope of existing data repositories and necessitates strict access control policies, severely hindering the pace of innovation 1172, 224]. Unfortunately, existing regulatory mechanisms for protecting sensitive patient data, such as the NIH's HIPAA guidelines, are often too slowly reactive to the fast-changing community perspective on biomedical data privacy. We need principled and effective computa-tional approaches that protect individual privacy while allowing researchers to leverage sensitive genomic or clinical data for science.
Next, even in settings where limited data access is not a primary concern, the sheer scale of emerging biomedical datasets often overwhelm many existing software tools for data analysis, requiring impractical runtime, storage, or memory resources. This difficulty often prompts researchers to use simpler yet less effective analysis tools, which likely render many important biomedical insights in the data inaccessible to researchers. Furthermore, the overwhelming computational burden of large-scale datasets restricts their utility for research labs without sufficient computational resources, which is detrimental to cultivating diverse perspectives in the field. We need efficient and accurate computational pipelines
for performing essential analysis tasks in biomedicine that gracefully scale with dataset size and use only a readily available amount of computational resources.
Lastly, the growing variety of biomedical datasets spanning different modalities of bio-logical systems (e.g., at the level of DNA, RNA, or protein) is helping researchers to gain a holistic understanding of biology. The majority of existing analysis tools are designed to process a single data type, requiring researchers to manually synthesize findings across different study settings, which is neither efficient nor effective for capturing weak but impor-tant signals that can be identified only by simultaneously analyzing multiple aspects of the biological system. We need principled computational tools that take multi-modal data from heterogeneous sources as input and extract insights into the underly-ing biological model that governs the similarities as well as differences between different data sources.
Taken together, these new computational challenges posed by the fast-accumulating biomedical data depict the increasing dependence of biomedical research on computer science and mathematics. A new wave of creative computational approaches is needed to fully tap into the emerging landscape of biomedical information to help accelerate scientific discoveries and their translation to medicine.
1.3
Contributions of This Research
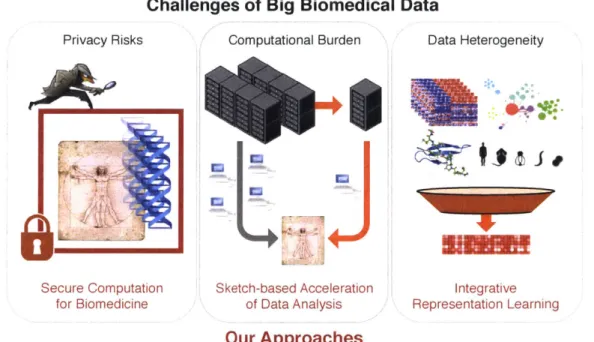
This thesis introduces a range of novel computational techniques to tackle the key scalability challenges of big biomedical data analysis across different areas of biomedicine, including medical genetics, pharmacology, single-cell genomics, and network biology. An overview of the approaches explored in this thesis are illustrated in Figure 1-1.
To address the privacy barrier associated with sharing sensitive biomedical data across different research entities, we develop secure multiparty computation pipelines based on mod-ern cryptographic tools, which enable researchers to compute over a dataset without seeing any of the underlying data values (akin to manipulating objects while blindfolded). By provably ensuring that the input data with potentially sensitive information is accessible only to the entity that provided the data, our secure pipelines aim to encourage
Privacy Risks
Secure Computat for Biomedicin
Challenges of Big Biomedical
Computational Burden
ion Sketch-based Acceleration
a of Data Analysis Data Data Heterogeneity Integrative Representation Learning Our Approaches
Figure 1-1: Overview of this thesis.
rative studies among multiple institutes and study participation of private individuals. We overcome the overwhelming computational burden of existing approaches, which have thus far limited the community adoption of these approaches, with novel techniques aimed at reducing the redundancy in computation. As a result, we obtain the first practical privacy-preserving pipelines for large-scale genentic studies (Secure GWAS) as well as computational drug discovery pipelines (Secure DTI).
To cope with the exponentially increasing scale of single-cell datasets, we explore how computational analyses could be performed based upon a compact summary (also called a 'sketch') of the dataset so as to vastly improve scalability while maintaining-or some-times even improving-the accuracy of the results. The key insight that we leverage is that large-scale single-cell measurements (e.g., including hundreds of thousands of cells) contain a considerable amount of redundancy, which could be reduced to gain significant computa-tional speedups without sacrificing the quality of results. Based on this insight, we develop a scalable algorithm (net-SNE) for visualizing high-dimensional single-cell measurements in two or three dimensions, which additionally achieves a useful property that it can be used to instantly visualize new data points in the context of existing visualizations, facilitating
knowledge transfer across multiple datasets. In a subsequent work, we directly address the question of how to best summarize a large-scale dataset with a small subset of cells. We introduce a geometric approach to single-cell data summarization (GeoSketch), which is extremely efficient and also enables accurate visualization, clustering, and batch effect correction with greatly accelerated performance. These results provide necessary computa-tional tools to broaden researchers' access to the emerging petabyte-scale data repositories of diverse single-cell experiments.
Finally, we tackle the multi-modal data analysis challenge in network biology by intro-ducing a new method (Mashup) for learning a low-dimensional vector representation of genes (or proteins) that succintly capture their interaction patterns across a variety of contexts. We demonstrate that our learned vectors can enhance the accuracy of a broad range of down-stream analysis tasks, including the inference of functional properties of genes, hierarchical clustering of genes based on their function, and genetic interaction prediction, compared to previous approaches of integrating heterogeneous datasets. Our followup work introduced a more advanced algorithm (clusDCA) for the function prediction task, where we additionally incorporate the semantic hierarchy of functional categories into the prediction procedure to gain significant improvements in accuracy, especially for functional categories where not many associated genes are known. These contributions enable biomedical researchers to jointly analyze a range of biological networks to gain more accurate insights into the bio-logical role of each gene or protein, and to further leverage that information to potentially derive biologically and clinically meaningful predictions.
1.4
Roadmap of Thesis
The remainder of this document is organized as follows. Chapter 2 provides introductory descriptions of different biomedical data analysis domains discussed in this thesis. Chapter 3 describes scalable secure computation pipelines for genome-wide association studies and pharmacological machine learning (Secure GWAS and Secure DTI). Chapter 4 describes scalable methods for analyzing large-scale single-cell datasets (net-SNE and GeoSketch). Chapter 5 describes scalable methods for integratively analyzing multiple biological networks
24
constructed from heterogeneous data sources (Mashup and clusDCA). Chapter 6 concludes the thesis with a brief summary of results and future directions.
Chapter 2
Background
This thesis introduces computational methods for data analysis tasks for a range of biomed-ical domains. To help orient the reader, introductory materials for these different areas of research are presented in this chapter.
2.1
Genome-wide Association Studies
A large portion of what makes us human is encoded in the human genome, which can be viewed as a sequence of around 3 billion 'nucleotides,' including adenines (A), cytosines (C), guanines (G), and thymines (T). When we compare the genomes of two different individ-uals, we typically find many spots, called 'genetic variants,' where the genetic code differs between the indviduals. The set of genetic letters one has at these variable locations within the genome is also called 'genotypes,' which intuitively characterize the genetic identity of a person. Such patterns of natural variation in genome across individuals provides an impor-tant source of information for teasing apart complex biological mechanisms underlying the human genetic code.
Since the introduction of next-generation sequencing (NGS) technologies, which enabled researchers to obtain genetic sequences of increasingly larger groups of individuals,
genome-wide association studies (GWAS) have been a key driving force in human genetics research
1265].s
GWAS describes an approach to experimental design where individual-level genetic data is collected from a group of study participants, and the genetic variants that arecor-Genotypes Phenotype
(e.g., has diabetes?)
T G C A Yes
TTC-G No
Individuals - - -
-T TA A Yes
Correated?
Figure 2-1: Illustration of GWAS.
related with observed traits of interest such as whether someone has a certain disease-also called 'phenotypes'-are identified through the statistical framework of hypothesis testing (e.g.,
X
2 test, Cochran-Armitage test, or likelihood ratio test). Genetic variants found to be associated with the disease under study can be further studied to gain insights into the eti-ology of disease or to derive clinical strategies for fighting the disease. Figure 2-1 illustrates the basic setup of GWAS.The key strength of GWAS lies in its genome-wide scale-it considers a comprehensive set of genetic variants that explain the biological differences among individuals in an agnostic manner. However, this wide scope comes at the cost of reduced statistical power. When a large number of hypotheses (i.e., whether each genetic variant is linked with phenotype) is tested, we run into the problem of false discovery; a seemingly significant association may have happened by random chance only because we are considering so many genetic variants simultaneously. To address this problem, it is standard procedure to apply correction methods that lead to more stringent thresholds for calling a finding statistically significant. However, much of the weak signals in the data (e.g., rare variants or weak associations) are washed out during this process and become even harder to identify. As such, it is crucial to amass datasets including sufficiently large number of individuals so that important biological signals in the genome can be detected. This is the key motivation behind ongoing international efforts to build biobanks of unprecedented scale.
A major challenge in GWAS lies in determining whether genetic variants that are sta-tistically associated with the phenotype are biologically meaningful or causal. For example,
confounding factors, such as age or sex, may cause the distribution of genotypes in the dataset to be skewed irrespective of the phenotype of interest, thus leading to spurious as-sociations. Although some of these factors can be easily recorded as meta-data during data collection and accounted for in the GWAS pipeline, more complex factors such as ancestry information can be difficult to ascertain in an explicit manner. In most GWAS experiments based on heterogeneous study cohorts, unsupervised correction for population stratification is considered a necessary step, where the most salient, high-level differences in genotypic data across individuals-captured via principal component analysis (PCA) [214] or linear mixed models (LMM) [1501-are ascribed to population stratification and controlled for in association tests. These additional measures increase the utility of GWAS by boosting the biological relevance of significant findings for the traits being studied.
2.2
Drug-Target Interaction Prediction
The goal of drug development is to find chemical compounds that interact with and alter the behavior of biomolecules in our body (e.g., proteins) in a way that helps combat disease and reduce suffering. Although high-throughput experimental technologies have been developed to enable reseachers to 'screen' (i.e., test the activity of) drug candidates at large scale, the space of interaction between millions of chemical compounds and tens of thousands of protein targets-also referred to as drug-target interaction (DTI)-is still too large to be exhaustively tested in a lab setting. To cope with this challenge, research labs typically rely on in sillico approaches for predicting DTIs in order to guide and prioritize lab experiments
[491.
At a high-level, the two main branches of in sillico methods for DTI prediction in-clude (i) molecular dynamics-based prediction, where the physical interaction between a drug compound and a protein are directly modeled at a molecular level, and (ii) ligand-based prediction, where the predictive relationship between more coarse chemical and structural properties of the molecules and the likelihood of interaction is learned from a database of a known set of interactions. Although the molecular dynamics approach enables de novo prediction for novel chemical compounds and protein targets that are poorly understood,
Drug Compounds Protein Targets Known OH Interactions OH/ H H 0? O OCH 3 / N/ - S Computational Models
Figure 2-2: Illustration of drug-target interaction (DTI) prediction.
it incurs overwhelming computational burden, typically requiring large-scale supercomput-ing resources. On the other hand, ligand-based prediction, though lacksupercomput-ing a direct physical basis for the resulting predictions, more effectively leverages information from known inter-actions and generates predictions for large sets of compounds or proteins at a time. The work presented in this thesis (in Section 3.3) is mainly concerned with the ligand-based-or more accurately a 'chemogenomic' [286]-approach where the properties of both the chemical compound and the protein molecule are leveraged for prediction.
Figure 2-2 shows an illustration of the DTI prediction problem. The known set of inter-actions between chemical compounds and protein targets can be represented as a bipartite graph with compounds on one side and proteins on the other with edges connecting drug-target pairs that interact. A key idea is to exploit the fact that drugs or proteins with similar structural or chemical properties will share similar interaction patterns to generalize the known set of interactions to previously uncharacterized ones with the help of predictive computational models. It is worth noting that this problem domain is closely related to the literature on recommender systems in data mining where products are recommended to users based on their previous associations with other products.
State-of-the-art approaches to DTI prediction includes matrix factorization-based and
network diffusion-based approaches. In the matrix case, the known set of interactions is represented as a partially observed matrix, which is provided as input to matrix completion methods that typically rely on estimating the low-rank structure of the underlying matrix. In the graph case, known interactions are used to construct a graph (e.g., Figure 2-2) and random-walk or diffusion methods are used to extract topological structure of the graph and use it to predict missing links in the graph. This is an area of active method development; for instance, a recent network-based approach [168] was able to achieve the state-of-the-art performance by integrating a variety of data sources via their graph representations, in-cluding databases of disease-disease, drug-disease, and drug-drug associations. More recent deep learning approaches [20, 59] to embed chemical structures, protein structures, or in-teraction graphs into latent feature vector space offer another exciting direction for future improvements in DTI prediction.
2.3
Single-Cell Transcriptomics
Proteins are the workhorses of cellular function, carrying out important biochemical reac-tions and making up a large fraction of structural components. When protein molecules are synthesized in a cell based on the information encoded in the genome, intermediary products called the messenger RNAs (mRNAs) are first produced based on the genome (process of
transcription), and proteins are subsequently produced based on the mRNA molecules
(pro-cess of translation). This two-step pro(pro-cess is also known as the central dogma in molecular biology.
Advances in high-throughput sequencing for DNA have led to the development of se-quencing technologies for characterizing these RNA molecules in cells. RNA sese-quencing datasets provide a snapshot of the dynamic state of cells under study, in contrast to the mostly static information offered by the genome. This technique allows researchers not only to determine whether a 'gene' (a genetic element corresponding to a particular biomolecule such as a protein) is active in the sample, but also to estimate the level of transcriptional activity of each gene. Such quantitative measures of transcriptional activity of individual genes provide a useful proxy for protein abundance in biological systems, since the latter is
Single-Cell Analysis
Single-Cell input
Bulk Analysis
Bulk RNA input
ur
-Each cell type has a distinct expression profile
Average gene expression from all cells
Reveals heterogeneity and subpopulation expression variability of thousands of cells Cellular heterogeneity masked
Figure 2-3: Illustration of single-cell RNA sequencing. Image from 10x Ge-nomics.*
significantly harder to experimentally determine in a high-throughput manner. The area of research concerned with extracting biological insights from high-throughput RNA measure-ments is known as transcriptomics.
Traditionally, RNA sequencing is performed based on a sample containing a large popula-tion of cells, commonly referred to as 'bulk' analysis. In this case, the resulting measurements of transcriptional activity can be thought of as an average taken over the population of cells. Recent advances in single-cell isolation techniques as well as better sequencing technolo-gies have led to single-cell RNA sequencing [95], where RNA measurements are taken at a
single-cell resolution. This new capability enables researchers to study the rich variability of
transcriptional activity within a population and to identify more fine-grain biological states of cells. A graphical illustration of single-cell RNA sequencing is provided in Figure 2-3.
Single-cell transcriptomics has led to numerous novel biological discoveries in a range of fields, including developmental biology, immunology, cancer biology, study of psychiatric disorders, among many others [95]. Emboldened by the wide success, academic consortia have been formed, such as the Human Cell Atlas (HCA) [219], to put together a compre-hensive map of single cells across different organ systems in human. Moreover, other 'omics'
*https://community.
lOxgenomics.com/t5/lOx-Blog/Single-Cell-RNA-Seq-An-Introductory-Overview-and-Tools-for/ba-p/547
technologies are currently being extended to the scale of single cells, including proteomics
[78j
and genetic perturbation techniques[77].
These ongoing developments suggest that single-cell datasets will have an increasingly important role in biomedicine in the future.2.4
Network Biology
Biological processes in our body are carried out through a sophisticated interplay of proteins and other biomolecules. This complex network of interactions can be mathematically rep-resented as a graph, where nodes represent individual biomolecules and edges represent an association between pairs of molecules. Although physical protein-protein interaction (PPI) networks are most widely studied, other forms of associations have also been studied as a graph, including genetic interactions (i.e., functional interaction of mutations in the genome), co-expression (i.e., correlated transcriptional activity), co-evolution, and co-occurrence in lit-erature. Furthermore, high-throughput experimental techniques for assessing the interaction between pairs of proteins have led to the construction of genome-scale interaction networks in diverse organisms, which are commonly referred to as the 'interactomes.' An example interactome of yeast from a seminal study by Costanzo et al. [62] is shown in Figure 2-4. The key insight driving the study of interactomes, also known as 'interactomics,' is that by inspecting the connectivity of these graphs in a top-down manner, one can learn a lot about how complex biological systems operate as well as the functional roles of individual biomolecules, which may lead to important clinical insights.
Many researchers have exploited the functional information encoded by the network struc-ture for diverse biological data analysis problems
163].
For example, one can predict the function of poorly understood proteins based on other proteins with known function that are closely located in the network. In addition, one can uncover the functional hierarchy of proteins in an unsupervised manner by analyzing the clustering patterns of the graph. Furthermore, interactomes serve as a useful guide for inferring which functional units (e.g., pathways or gene modules) explain downstream patterns that are observed at the level of in-dividual genes; in particular, this approach has been used for cancer mutation analysis[157]
as well as functional analysis of genome-wide association study results [106]. It is worthnot-Mitochondris Ribosome & %.. p' .
Las* n.Metabolism & amino acid biosynthesis NA Secretion & transciption& glycosytatdon Nucleavi cytoplasmmlc * * . transporransport
Mitosis & cni DiA repIcatIafn segrCgatioii & rep*ir
Figure 2-4: Visualization of the yeast interactome. Genetic interaction-based
as-sociation network from Costanzo et al.
[621
Groups of genes enriched for specific biological processes are highlighted.ing that the increasing scale of biological datasets are enabling researchers to characterize more fine-grain interactomes for specific biological contexts, including different tissues
[1061
and more recently, single cells [671. However, given the relative incompleteness and noisy
nature of the current human interactome datasets, the biomedical community needs future efforts in enhancing the quality of human interactome and developing more effective compu-tational algorithms for teasing apart useful biological signals from noise from the large-scale and heterogeneous interactome datasets.
Chapter 3
Privacy-Preserving Biomedicine at Scale
3.1
Overview
Collaboration is essential to modern translational genomics research. The sheer amount of genomic and clinical data required to better understand human genetics and its impact on health and disease requires data sharing across many different labs and institutes. Although several national and international collaborative research partnerships have formed to facili-tate the sharing of biomedical datasets [246, 96, 50, 86, 101], the restricted scope and strict access control policies of existing data repositories severely hinder the amount and pace of innovation.
A key challenge in broadening the scope of data sharing is the sensitive nature of biomed-ical information
[173].
Due to the growing scale of patient data in scientific studies, a single data breach can now leak genomic and other health-related information on millions of individ-uals, which may put the affected individuals at risk for genetic discrimination for employment or insurance, or unwanted disclosure of their biological family, medical history or sensitive disease status. The potential scope of such harm extends to the descendants or relatives of the affected individuals as they share much of their genetic biology. Moreover, unlike user accounts and passwords routinely leaked by IT companies, one's genetic information cannot be changed at will--once it is leaked, it stays leaked.A rapidly increasing number of studies demonstrate that many datasets, previously con-sidered safe to release, actually leak highly sensitive information even after taking preventive
measures
[118,
232, 109, 112, 891. Unfortunately, existing regulatory mechanisms for protect-ing sensitive patient data, such as the NIH's HIPAA guidelines, are often too slowly reactive to the fast-changing landscape of biomedical data privacy. Given the ongoing explosion of genomic and clinical data, data-driven biomedical research urgently needs principled and effective approaches to protect individual privacy.Modern cryptography techniques, including secure computation frameworks [64, 32, 2871, offer technological solutions to growing privacy concerns by allowing researchers to extract insights from sensitive data without directly accessing the raw data. Privacy-preserving data analysis pipelines hold great potential to enable secure data sharing and collaborative science among researchers, which has led to key method development efforts in the field such as the iDASH workshops [1311. However, community adoption of these cryptographic tools has been limited, due to their overwhelming computation burden for complex, large-scale data analysis. For example, a vanilla MPC protocol for a full genome-wide association study (described in Section 3.2), including essential population stratification analysis, requires years of computation to process a million genomes, which is not feasible for most study settings. Other approaches to secure computation, such as homomorphic encryption (HE) and Intel's Software Guard Extensions (SGX), similarly suffer from limited scalability; HE generally incurs greater cryptographic overhead than MPC for complex computations, and SGX supports highly limited memory while providing substantially weaker privacy.
This chapter introduces techniques for achieving practical privacy-preserving analysis for two essential biomedical applications: genome-wide association studies and drug-target
interaction prediction. These results provide valuable insights for building privacy-preserving
biomedical analysis protocols with practical computational requirements.
3.2
Secure Genome-wide Association Studies
3.2.1
Introduction
Genome-wide association studies (GWAS) have long been a major driver of genetics research. They aim to identify genetic variants that are statistically correlated with phenotypes of
36
interest (e.g., disease status). Analyzing a large number of individuals in a GWAS is critical for detecting weak, yet important genetic signals, such as rare variants or those with small effect sizes
[125,
185]. However, privacy concerns[35,
109, 232, 1121 have stymied such large-scale studies by discouraging individuals and institutions from sharing their genomes[224,
172] and necessitating strict access-control policies for the amassed datasets, which limit their utility.
Modern cryptography offers a new paradigm for genetics research we call secure genome
crowdsourcing, where the input data for population-based studies like GWAS are massively
pooled from private individuals or individual entities while hiding the sensitive information (i.e., genotypes and phenotypes) from any entity other than the original data owners. For example, secure multiparty computation (MPC) frameworks [64] enable researchers to col-laboratively perform analyses over securely shared data without having direct access to the underlying input. The confidentiality of input data guaranteed by such frameworks would greatly encourage genomic data sharing. Moreover, unlike the current practice of entrusting a single entity (e.g., a biobank [246, 96, 50]) with the raw data, a breach or corruption of a single party-an increasingly probable event in an era where companies' sensitive user data are routinely leaked in bulk-no longer compromises the privacy of study participants.
Despite their promises, existing proposals for securely performing GWAS based on cryp-tographic tools like MPC [131, 133, 165, 270, 61, 24, 28] are too limited to enable secure genome crowdsourcing in practice; they either consider vastly simplified versions of the task or require infeasible amounts of computational resources for modern datasets with a large number of individuals (e.g., many years of computation or petabytes of data; Section 3.5.4). For example, recent work [127], which introduces privacy-preserving rare variant analysis based on a type of MPC technique known as garbled circuits [287], is limited to simple Boolean operations and is not applicable to large-scale GWAS, as noted in their work.
A major computational bottleneck for secure GWAS is identifying population structure within the study cohort, which can cause spurious associations that are linked to inter-population differences rather than a true biological signal
[901.
A widely-used procedure for accounting for such confounding is to use principal component analysis (PCA) to capture broad patterns of genetic variation in the data [214]. The top principal components, whichare thought to be representative of population-level differences among the individuals, are included as covariates in the subsequent association tests to correct for bias. However, performing PCA on very large matrices is challenging for secure computation, which, to our knowledge, has never been successfully addressed. This barrier is mainly due to the iterative nature of PCA, which significantly increases the communication cost and the overall complexity of the computation. In addition, PCA requires computing over fractional values with sufficient precision. This introduces nontrivial overhead to most existing cryptographic frameworks which are inherently restricted to integer operations. Supporting computations over fractional values not only increases the size of the data representation, but also increases the complexity of the basic underlying operations such as multiplication and division. Here, we present the first secure multiparty computation protocol for GWAS that includes both quality control and population stratification correction while achieving practical performance at scale for even a million individuals.
3.2.2
Our Approach: Secure GWAS
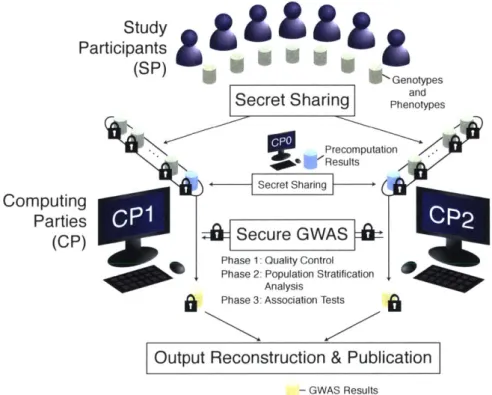
An overview of our pipeline is provided in Figure 3-1. Our protocol has two types of enti-ties: study participants (SPs) and computing parties (CPs). SPs refer to private individuals, institutions, or intermediary data custodians that own the genomes and phenotypes to be collectively analyzed for the study. CPs consist of three independent parties with appropriate computing resources (CPO, CP1, and CP2) that cooperatively carry out the GWAS
computa-tion. We envision academic research groups, consortia, or relevant government agencies (e.g., NIH) to play these roles. At the beginning of our protocol, each SP securely shares their data with CP1 and CP2 using a cryptographic technique called secret sharing
1171.
Next, CP1 and CP2 jointly execute an interactive protocol to perform GWAS over the secret shareswithout learning any information about the underlying data. During this step, precomputed values from CPO, which are independent of the data from the SPs, is used to greatly speed up the process. Importantly, CPO does not see the input and is involved only during prepro-cessing. Lastly, CP1 and CP2 combine their results to reconstruct the final GWAS statistics
and publish them. A complete protocol description is provided in Section 3.4.
Importantly, the total communication complexity of our protocol (i.e., the total amount
Pe Computing Parties (CP) rticipant (PGenotypes and
Secret Sharing Phenotypes
-. Precomputation
* Resuts
Secret Sharing
Secure GWAS
Phase 1: Quality Control Phase 2: Population Stratification
Analysis Phase 3: Association Tests
Output Reconstruction & Publication - GWAS Results
Figure 3-1: Overview of our secure GWAS pipeline. Study participants (private individuals or institutes) secretly share their genotypes and phenotypes with computing parties (research groups or government agencies), denoted CP1 and CP2, who jointly
carry out our secure GWAS protocol to obtain association statistics without revealing the underlying data to any party involved. An auxiliary computing party (CPo) performs input-independent precomputation to greatly speed up the main computation.
of data transferred between the CPs) scales linearly in the number of individuals (n) and the number of variants (m) for both the precomputation and the main computation phases after initial data sharing. In contrast, directly applying state-of-the-art MPC frameworks [25, 71, 136] leads to quadratic communication with large multiplicative constants, which is vastly impractical when both n and m are close to a million. This is primarily due to the fact that existing frameworks strictly adhere to a modular execution of the computation purely expressed in terms of elementary additions and multiplications; alternatively, refactoring the desired computation can lead to significant performance enhancements as we show in our work.
We introduce several key technical tools that enable our efficient protocol. First, we generalize a core MPC technique known as Beaver multiplication triples [16], which was initially developed for secure multiplication, to efficiently evaluate arithmetic circuits. Our generalized method enables efficient protocols for not only matrix multiplication, but also exponentiation and iterative algorithms with extensive data reuse patterns, all of which fea-ture prominently in secure GWAS. Second, we leverage random projection techniques [1111, which have been shown to be effective for other genomic analyses
[921,
to reduce the task of performing PCA on the large genotype matrix (in population stratification analysis) to factoring a small constant-sized matrix. Third, we employ cryptographic pseudorandom generators (PRGs) to greatly reduce the overall communication cost; when a CP needs to obtain a sequence of random numbers sampled by another CP, which constitutes a signifi-cant portion of our protocol, both parties simply derive the numbers from the shared PRG non-interactively. Lastly, we carefully restructure the GWAS computation such that each intermediate result (which requires the CPs to communicate a message of the same size) is of size linear in the input dimensions (n and m). We provide a comprehensive description and analysis of our techniques in Section 3.4.3.2.3
Methods
Secret sharing reviewSecret sharing
[17]
allows multiple parties to collectively represent a private value that can be revealed if a prespecified number of parties combine their information, but remains hidden otherwise. To illustrate, imagine an integer x that represents the genotype of an individual at a specific genomic locus. The value of x can be secretly shared with two researchers Alice and Bob by giving Alice a random number r and Bob x - r modulo a prime q, which perfectly hides x if r is uniformly chosen from the integers modulo q. While the information about x is encoded in the two shares without loss, either Alice or Bob alone does not learn anything about x. Using this technique, private individuals can freely contribute their genomes to the computing parties in our GWAS protocol, without giving anyone access to the raw data.Secure multiparty computation review
Multiparty computation (MPC) techniques based on secret sharing
[64]
enable indirect, privacy-preserving computation over the hidden input. For example, secure addition of two secretly shared numbers x and y can be performed by having both Alice and Bob add their individual shares for x and y. The new shares represent a secret sharing of x +y, which is the desired computation result. Secure building block protocols for more complicated operations (e.g., multiplication, division) are similarly defined, albeit with more advanced techniques that require certain messages (a sequence of numbers) to be exchanged between Alice and Bob. By composing these protocols, arbitrary computation over the private input-even GWAS-can be carried out while keeping the input data private throughout.Our MPC techniques for scaling secure GWAS
The key technical hurdle in applying secure MPC in practice has been its lack of scalability. The cost of communication between Alice and Bob quickly becomes impractical as the size of the input data grows and the desired computation becomes more complex. In particu-lar, principal component analysis (PCA) is a standard procedure for GWAS that incurs an overwhelming communication burden for a large input matrix (e.g., a million in each