g
(LIBRARIES)
C9>3,
HD28
.M414Dynamic
Multidrug
Therapies
for
HIV:
A
Control
Theoretic
Approach
Lawrence
M.
Wein,
Stefanos
Zenios,
and
Martin
A.
Nowak
MASSACHUSETTSINSTITUTE
J/
^23
1996Dynamic
Multidrug
Therapies
for
HIV:
A
Control Theoretic
Approach
Lawrence
M.
Wein
Sloan School of
Management,
M.I.T.Cambridge,
MA
02139
Stefanos Zenios
Operations Research Center, M.I.T.
Cambridge,
MA
02139
Martin
A.Nowak
Department
of Zoology, University ofOxford
Oxford
0X1
3PS,UK
ABSTRACT
Motivated by the inability of current drug treatment to provide long-term benefit to
HIV-infected individuals,we
deriveHIV
therapeutic strategiesby
formulatingand
analyzing amathematical
control problem.The model
tracks thedynamics
of uninfectedand
infectedCD4
+ cellsand
free virus,and
allows the virus tomutate
into various strains.At
each point in time, several different therapeutic options are available,where
each option correspondsto a
combination
of reverse transcriptase inhibitors.The
controller observes the individual's current statusand
choosesamong
the therapeutic options in adynamic
fashion in order tominimize" the total viral load.
Our
initial numerical results suggest thatdynamic
therapieshave the potential to significantly
outperform
the static protocols that are currently in use;by
anticipatingand
responding to the disease progression, thedynamic
strategy reduces thetotal free virus, increases the uninfected
CD4
+ count,and
delays theemergence
ofdrug-resistant strains.
December
1995
1.
Introduction
Optimal
treatment of thehuman
immunodeficiency
virus of type 1(HIV)
infection isthe subject of intense research activity.
Rapid
progress hasbeen
made
in thedevelopment
and
testing ofanti-HIV
therapeutic agents, resulting in the approval by theFood
and
Drug
Administration of fivereverse transcriptase
(RT)
inhibitors(AZT.
ddl,ddC, d4T,
3TC)
and
one
protease inhibitor (saquinavir).These
potent drugs inhibit viral replicationand
lead toa rapid decline in viral
abundance,
often within days after starting therapy. Unfortunately, these drugshave
had
only limited success in delaying the onset ofAIDS:
continual viral replication ofHIV,
together with the high errorrate of reverse transcription ofviralRNA
intoDNA.
leads to theemergence
ofdrug-resistant virus strains, typically withinweeks
ormonths
(depending
upon
the drug) of treatment initiation(McLeod
&
Hammer.
199*2;Lagakos
etaL. 1993; Volberding ef aL. 1994:
Wei
et a/.. 1995;Schuurman
et aL. 1995). Moreover, multidrug resistance hasbeen
documented
in combinations ofRT
inhibitors (Larder et aL.1993;
Richman
et ah. 1994; Shafer et aL. 1994, 1995;Kojima
et ai., 1995)and
combinationsof protease inhibitors
(Condra
et aL. 1995). although Larder et a!. (1995)have
recentlyshown
that theAZT-3TC
combination
is able to sustain in vivo antiviral effects for at least24 weeks.
It
would appear
that increased effectiveness ofHIV
therapy couldbe
achievedby
developingdynamic
multidrug approaches,where
thecombination
of drugs receivedby
a patient changes over time in response to the disease progression (e.g., currentC4
+ count,viral load,
mix
of viral strains). In thispaper
we
mathematically
model
thedynamic
multipledrug
therapyproblem
asan
optimal controlproblem
that canbe
informally described as follows: choose an optimalmix
of drugs at each point in time in order tominimize
the total viral load oversome
time horizon (e.g..one
year) subject to atoxicity constrainton
thedrug
regimen.Several
mathematical
models have been
developed that incorporate theeffects ofther-apy on
an HIV-infected individuals. In a series of papers (Perelson, 1989; Perelson et al., 1993; Kirschner&
Perelson, 1994; Kirschner&
Webb,
1995a; Kirschner et al, 1995), Perelsonand
Kirschnerand
their colleagueshave
studied the timing, frequencyand
intensity ofAZT
treatment.Agur
(1989) focuseson
the optimal tradeoffbetween
the toxicityand
efficacy ofAZT.
The
mathematical
models
considered in these studiesdo
not allow for virus mutation.Nowak
et al. (1901),McLean
t
Nowak
(1992),Nowak
k
May
(1993), Frostk
McLean
(1994)and
Kirschner&
Webb
(1995b) analyze descriptive (asopposed
to optimization)models
forthe competitive interaction of AZT-sensitive
and
AZT-resistant strains ofHIV;
the latterthreepapers also include numerical simulations of alternating
and/or
combination therapies.Our
deterministic control problem, which is described in Section 2, considers a finitenumber
of virus strains, or quasispecies,and
allows mutationsfrom one
strain to another.A
finitenumber
of therapeutic options are allowed,where
each option consists of thesi-multaneous
application ofone
ormore
RT
inhibitors.The
model
incorporates uninfectedCD4
+ T-cells,and
infected
CD4
+ cellsand
infectious free virus associated with each virusstrain; in this context, the
RT
inhibitors prevent the free virusfrom
successfully infecting uninfectedCD4
+ cells.Each
drug option has a different efficacy against each virus strain,thereby allowing for
complex
drug-virus interactions.Because
of the high dimensionality of the control problem,we
resort toapproximation
methods
in Section 3;more
specifically, perturbationmethods
and
the policyimprovement
algorithm areused to derive a closedform
dynamic
policy. Severalother typesoftherapeuticapproaches, such as protease inhibitors
and
the reconstitution of theimmune
system, are considered in Section 4. Resultsfrom
numerical simulations are reported in Section 5and
discussed in Section 6.Concluding remarks
canbe
found in Section 7.2.
Problem
Formulation
Our
mathematical
model
incorporates / different strains ofHIV.
For i=
1. . . . ,/, letyi(t) be the density of
CD4
+ cells infected by strain i at time r,and
let u,-(£) denote the density of infectious free virus of strain i; noninfectious virions are ignored in our model. Ifwe
define x(t) tobe
the density of uninfectedCD4
+ cells at time t, then the state of thesystem
at time t is givenby
(x(t), yi(t),...
,yi(t), V\{t), .. . ,vj(t)),
which
isdenoted by
(x(t).yi(t).vt(t)).
The
controller hasJ
therapeutic options at time t,where
each option corresponds toa combination of
RT
inhibitors. For example, j=
1might
representAZT
used with ddland
combination j'=
2
might
be
AZT.
ddC
and d4T.
Our
control variables are dj(t).which
is the
amount
of option j to use at time t.We
assume
that each virus strain has itsown
infectivity rate,
denoted
by
3,.which
is the rate that it infects uninfectedCD4
+ cells; thereason for the "tilde" will
become
clear in the next section.The
RT
inhibitors reduce virus infectivity in the followingmanner.
For i=
1....,/and
j=
1,...,J, letp
3i denote the
efficacy of drug
combination
j in blockingnew
infectionsby
virus strain i.Under
a genericdrug policy dj(t), the infectivity of virus i is $i[l
—
J2j Pj>dj{t)]:we
assume
that the valuesof pji
and
the toxicity parameters in equation (4) are chosen so that the infectivity of eachstrain is nonnegative
under
all feasible therapeutic strategies.The
dynamics
of oursystem
are describedby
the following set of ordinary differentialequations:
x(t)
=
A-
(f,+
£
ft«j(0[l-
X>*
d
i(*)])*(*). (1) >=1 .7=1J
m
=
(E
9wW*)[i
-
E/wM')Lk>)
-
o.-y.-(*),(2)
fc=l 3=1
bi{t)
=
iriyi(t)-[ki
+
Pix(t)]vi(t). (3)and
each ofthese potential infections leads to a reduction in free virus.The
rate of successful infections bystrain i is /?,-[l—
Z)j=] Pjidj(t)]vi(t)x(t),and
theseinfections cause asimultaneousdecline in uninfected cells x(t)
and
rise in infected cells y,(i).The
mutation
rate gtj in (2) is the fraction of reverse transcriptions of strain i that result in a cell infectedby
strain j. Hence, J2j=i <7«j=
1an
d the diagonal terms of themutation
matrix are close toone
invalue, while the off-diagonal terms are nearly zero. Strain i replicates at rate 7r, after it has
infected a
CD4
+ cell,and
thus free virus of strain i isproduced
at rate 7T,-y,-(i); an alternativeinterpretation is that Tr
t
/a
t virions areproduced by
a lytic burst during the death of aninfected cell. Notice that thequantities pJt-, qi
3
and
7rj can capture awide
range of drug-virusinteractions, including both synergistic
and
antagonistic effects of combination therapy, asobserved by St. Clair et a/. (1991)
and
Larder ef a/. (1995),and
surveyedby
Wilson
and
Hirsch (1995).Uninfected
CD4
+ cells increase because of (exponential) proliferation in peripheraltissue
compartments
(e.g., secondarylymphoid
organs)and/or
(linear) productionfrom
apool of precursors (e.g., the thymus). For simplicity,
we
assume
a constant production rate A. In addition,we
let [t,a
t
and
kt denote the respective natural death rates of uninfectedCD4
+ cells, cells infectedby
strain iand
free virus of strain i.Because
theprimary
focus of thispaper
ison
therapeutic regimensand
noton
natural disease progression,we
purposelydo
not incorporate thehuman immune
response into themodel. Hence, the
mutants
areassumed
to escapefrom
the drugs, notfrom
theimmune
response.
The
model
also ignores latently infected cells; althoughmost plasma
viruscomes
from
actively infected cells(Ho
et ai, 1995;Wei
et a/.), this does not imply that latently infected cells areunimportant
for theemergence
ofdrug resistance. Finally, ourmodel
doesnot incorporate
new
datafrom
Shaw
(1995).which
suggests that thelymph
system
is theWe
impose
the toxicityand
nonnegativity constraints£^(0<i.
(4)3=1
dj(t)
>
0. (5)where
t3 is a (normalized)
measure
of the toxicity per unit time ofdrug
combination j.Hence,
we
assume
that toxicity is additive across drugs,and
that the toxicity threshold isconstant overtime (and is takento
be
unity, without loss ofgenerality).However,
because thepolicies that
emerge from
our analysis typically use onlyone
therapeutic option at each pointin time, the additivity
assumption
in (4) is innocuous. Moreover, thedynamic
policy canbe
easily
adapted
to themore
realistic settingwhere
the toxicity threshold is an exogenouslyspecified function of either time (e.g.. six
months
of intense therapy is alternated with sixmonths
of light therapy) or the severity of the patient's side effects:we
propose that thedrug
combination
dictatedby
the index policy in the next sectionbe
employed
at each pointin time, but the dosage should
be
altered accordingly.Let
T
be
thetime
horizon.Then
themathematical
controlproblem
is to choose the controls {dj(t),t>
0} tominimize
L
T5>(*)d
(6) t=l subject to (l)-(5).Our
controlproblem assumes
that the decisionmaker,
in choosing dj(t) at time t, canobserve the current state (x(t),yl(t).vl{t)).
Although
recent technologymakes
it possibleto quantify virus load,
CD4
+ cell countsand
virus-infected cells in the blood of infected individuals, these techniques are not currently available for day-to-day treatment ofHIV-infected patients at large. Moreover, quantification of virus load
and
infected cells in thelymph
system
is evenmore
complicated.A
more
realistic controlproblem
for currentonly the total
CD4
+ count x(t)+
YlUi Vi(t)an
^ the total vir^
l°a<iHi=i
l';(0)-The
controlproblem
(l)-(6) thenbecomes
a nonlinear filtering problem; although partial observations greatly complicate the problem, reasonablygood
policies (e.g., the linearizedKalman
filteron page
161 of Elliott,Aggoun
and
Morse, 1994) canbe
derived. However, becausewe
are primarily interested in the potential value of
employing dynamic
multidrug therapies,we
do
not pursue this line of inquiryand
hereafterassume
that the controller can observe(x(t),yi(t),Vi(t)} at time t.
3.
Analysis
The
controlproblem
(l)-(6) does notappear
toadmit
a closedform
solution.More-over, standard numerical techniques based
on
Pontryagin'smaximum
principle (Pontrvaginct ai. 1962) cannot solve a
problem
of realistic size (e.g., five drug optionsand
30 virus strains). Hence,we
resort to anapproximate
method, which employs
perturbationmethods
in conjunction with ideas
from
dynamic
programming.
3.1.
Overview.
LetV(x.y
t.vt,t) denote the cost incurred (as given in (6))from
time t to time
T
under
the optimal policy, given that the state of thesystem
at time t is(x,yi,Vi). For ease of notation,
we
suppress thedependence
ofV
on
its arguments.Then
the
dynamic
programming
optimality equation is (Bellman. 1957)dv
'f^dv,
„
. . ,\ . fdV
+
mm
< -7—
{^GfJ}l d.r j 1=1 J=l J (<) fc=l j=\where
Q
denotes the set of admissible controls that satisfy (4)-(5).Our
basicapproach
is as follows:we
use asymptoticmethods
to obtain closedform approximate
expressions for the(2I+l)-dimensional process {(x(t),yi(t),Vi(t)}
under
a static control policy (i.e., dj(t)=
d3
for all t).
From
these expressions,we
derive a. closedform
expression forV
which has thesame
interpretation as theV
in (7), except that the (approximate) cost isunder
the static policy, not the optimal policy.Then
one
iteration of the policyimprovement
algorithm(Howard.
1960) is performed:we
differentiateourapproximate
closedform
expression forV
with respect to x
and
yt. substitute these derivatives into the right side of (7)and perform
the
embedded
minimization in this equation.This
approach
yields a closedform
dynamic
drug
control policy: it specifieshow
much
of each drugcombination
to use at each point in time in terms of the current state, the current time,and
all theproblem
parameters. If our closedform
expression forV
was
exact, thendynamic
programming
theorywould imply
that our proposed policywas
better(yields a lower cost in (6)) than the static policy (in fact, if our expression for I'
was
exact then repeated policy
improvement
iterationswould
generate a sequence of policies that converges to the optimal policy).However,
our expression is not exact,and
we
cannotdraw
this conclusion. Nevertheless, this philosophy (finding anapproximate
value forV
and
performingone
iteration of the policyimprovement
algorithm) hasbeen
used withconsiderable success in designing
dynamic
call acceptance/rejection protocols for stochasticmodels
of telephone traffic (e.g., Ottand
Krishnan, 1985; Key. 1990).3.2.
The
Optimal
Static Policy.Because
ourproposed
dynamic
policy takes astatic policy as its starting point, it is naturalto
employ
the optimal static policy.The
staticoptimization
problem
is givenby
the following nonlinearprogram:
r
minimize,..,,,.,,,.^
^
v, (8)!=1
bject to A
-(fi
+
Yl
&«i[l"
EM;])-
r=
°-(9) i=i j=i
(E
q*hvk[l -J^Pjkdifjx
-
a
iyi=
0. (10)fc=i j=i
£^<1,
(12)dj>0.
(13)We
havebeen
unable to find a closedform
solution to (8)-(13); however, standard numerical techniques (e.g., Avriel. 1976) can be used to solve this problem. If the additivity con-straint (12) is not realistic thenone
canchange
constraint (13) to d3=
ortJ
for all j; thisnew
problem
canbe
solvedby
directenumeration
of allJ
+
1 feasible solutions.Let d* denote the optimal static policy that solves (8)-( 13).
Under
this policy, theinfectivity of virus i is /^J,,
where d
t=
1—
J2j=iPji^j-3.3.
Asymptotic
Analysis.
In this subsectionwe
find a closedform approximate
expression for the
system
trajectoryunder
the optimal static policy. Consider equations (1)-(3). with J, taking the placeof 1
—
5l/=i Pjidj(t).Keeping
inmind
thatwe
will usethe solulion to these equations to derive V. let us consider the initial conditions (in this subsection, s is a generic time indexand
t denotes a specific point in time)x(t)
=
x, yi{t)=
yi Vi{t)=
Vi. (14)To
perform
the perturbation analysis,we
introduce theparameter
e.Without
loss of generality, let usassume
that @\=
rnin{i<,<j} j3t.Then we
let e=
/?iand
define&,
=
-
(15)f
Although
0!=
1,we
retain thisparameter
in the model.The
asymptotic analysis is basedon
theassumption
that e is small in value (i.e.,much
less than one). Typical values in the literature for the infectivityparameter
$
are roughly10-5; hence, the approximation should
perform
well.We
assume
that our solution is of the form:x(s)
=
XM(s)
+
ex^(s). y>(s)=
y\0)(s)+
ey{ t l \s). Vi(s)-
v\ 0) (s)+
er' 1 '^). (16) 8Although
we
could defineand
solve for higher order terms,just deriving the first order termsis quite
cumbersome,
at least without acomputer.
We
use (15) to replace (3tby
e/?tand
use (16) to substitute in for the
system
state in equations (l)-(3)and
(14). For i=
0,1.we
collect terms of order e' to solve for theunknown
processeson
the right side of (16). Collecting the constant (i.e., e°) terms yields the followingsystem
of differential equations:x{0)
(s)
=
\-iix
{0]{s). (17)y\ 0)
(s)
=
-*iy\
0)(s), (is)ii°\s)
=
nyl
0)(3)-
kiV^is), (19)subject to the initial conditions
,-<°>(0
=
.r. y\°\t)=
yt
$\t)
=
», (20)It is
worth
noting that themathematically
perturbedsystem
(17)-(20) corresponds precisely to the physical perturbationperformed
when
giving potentRT
inhibitors. In Section 4,we
usedata
from
Wei
et ai.,Ho
et al.and
Perelson et al. (1995),who
used variousRT
inhibitorsand
protease inhibitors to perturb the system, to estimate values for themodel
parameters.The
solution to equations (17)-(20) isz(o)(3)
= A +
(x
_
A)
e-M(.-*)) (21)V?\s)
=
Vi*-ai{~
t\
(22) „<*»(,)=
JEiW_
e-a,(.^)+
{v._
J™_
)e-M*-*). (23) k t—
a, kt—
a;The
order e terms lead to the followingsystem
of differential equations:I
x(%) =
-
M
xW(
a)-x(°>(
a)j:M»}
0)(-)i (24) »} i)(-)=x
(o,(*)(x:««A^f
,(*))-
a
*»i 1) (*). (25) 9v\'\s)
=
r
iyl%)
-
kivPia)
-
i3tx^(s)vl
0)
(s). (26)
subject to the initial conditions
.r<1)
(0
=
0,yV(t)
=
u{1)
(i)
=
0. (27)Now
we
substitute the solution (21)-(23) into (24)-(26)and
solveequations (24)-(27)sequen-tially.
The
solution is given inAppendix
A.3.4.
The
Proposed
Dynamic
Policy.With
the solution to the asymptoticsys-tems
(17)-(20)and
(24)-(27)in hand,we
can estimatethe value functionV
by
Jt 5Zf_1(f,- {s)
+
ev\ (s))ds. This integration is carried out in
Appendix
B.The
proposed
solution is greatlysimplified if
we assume
that the value function is independent of the time horizon. Inprac-tice, the time horizon
T
is very large (i.e.. in years) in relation to the time scale of thesystems dynamics,
which change on
a daily basis(Ho
ef al..Wei
et a/.): hence, this is a natural assumption. Consequently,we
setT =
ocand
/=
in (46) to obtain a value function (andhence
a policy) that is independent of the time horizon; this approximate,stationary value function is denoted by
V
c. to distinguish itfrom
the value function in (7).Differentiating (46) with respect to x
and
yt yields (withT
=
ooand
t=
0)WctUt^-l)!*)^^),
(28)+
e Wij3./_^_
+
x~
A//' \ (j.y-
Mil
-
-'\otikiH (ai
+
fi)(ki+
fi)J I 'J=l otjkj Ja
(29)
%
CtikiNow
we
can substitute these derivatives into (7)and perform
the minimization. Be-cause the function tobe minimized
is linear in our controls dj(t), the solution d*(t) isone
ofthe (at
most J
+
1)extreme
points ofthe constraint set (4)-(5),and
canbe found
in closedform. Let us define the
dynamic
quantitiesI Ci{t)
=
0Mt)
10
/Ki quel,
_ J_
y.
*jqijdi\ /7T/[^A(Qf+
A-;+
//)+
Q
fAy(?,/.;(Q-
yf(0]_
fv(Q \^
";A, fc,^
ajkj '\ ctiki(cu+
fi)(kt+
(i)k +
(i'Then
theproposed
policy is: applyno
therapy (i.e., d^(t)=
forj=
1J
) if(30)
max
<
0: (31){l<j<J} Tj
otherwise, use
drug
combination j' (i.e.. d".(t)=
t~, , d~(t)=
for j^
j"),where
eLiPj.-c»(0 /QOx
j
=
argmax^^^jj
. (32)We
conclude this subsection with several remarks.The
proposed
therapeutic strategy is adynamic
indexpolicy,where drug combination
j has the
dynamic
indexH
t=] PjiCi(t)/Tj,and
at each point in time the policy uses the drug combination that possesses the largest index.The
quantity ct(t) essentially represents themarginal increase in the total future viral load if
we
letone
more
CI)4+ cell get infected byvirus strain i at time /.
The
index fordrug combination
j iscomputed
by
weighting thedynamic
marginal cost for each virusby
the efficiency ofdrug
j for that virus,summing
up
over thevirus strains,
and
dividingby
thedrug
combination's toxicity. Hence, ourdynamic
policy uses information
on
the effectiveness of eachdrug
against each virus, the current potential cost (in terms of total future viral load) of anew
infectionby
each virus strain,and
the toxicity of eachdrug
combination,and summarizes
this information in a succinctmanner.
.An
importantadvantage
of index policies is their ease of use: the complexity ofthe derivation
and
implementation of an index policy is independent of theproblem
size; hence, policy (30)-(32) can easilybe
derived for aproblem
with 20drug
combinationsand
150 virus strains.Although
this policy is not the optimal solution toproblem
(l)-(6). theoptimal solution to
dynamic
resource allocationproblems
is often characterizedby
indexpolicies (e.g.. Gittins. 1989).
Notice that if
we
took e=
in (30) then the policywould be independent
of xand
?/,-,whereas
theproposed
policydepends
on
the entire (2/ -+- l)-dimensionalsystem
state; thissuggests that incorporating only the e° terms leads to a rather crude policy. Also, one ofthe
drug combinations
would
alwaysbe
administered (i.e., inequality (31) never holds) if e=
0.Inquality (31)
was
never satisfied inany
ofour numerical runs: the proposeddynamic
policyalways used
one
of the drug combinations.It is possible to
implement
furtheriterations of thepolicyimprovement
algortihm. Let us denote theproposed
policyin (30)-(32)by
eQ (t); although this control is state-dependent, it canbe
expressed solely as a function of time because thesystem
is deterministic. Ifwe
define d
t
(t)
=
1—
J2j=iPjidj \t) then this quantity canbe
used as our starting policy for the next policyimprovement
iteration. Turning to the asymptotic analysis,we
observethat the e°-order
system
(17)-(20)and
its solution (21)-(23) are independent of the control.If
we
substitute d] (t) in for dt in equations (24)-(25). then theeorder system
(24)-(27)is still a set of linear differential equations thai can be easily solved numerically using the
matrix exponential
(Golub and
Van
Loan. 1989).Then we
can carry out the calculations inAppendix
B
and
equations (28)-(29)on
a computer,and perform
the minimization in (7)to get a
new
policy d} (t).
Of
course, higher order terms in theasymptoic
expansion canalso
be
performed
relatively easily with a computer,and
it is conceivable that, withenough
higher order termsand enough
policy iterations (typically, only a handful of the latter is required to get close to optimality). such a procedurewould
generate a policy that is veryclose to optimal.
However,
because theproposed
policy in (30)-(32)performed
well in our numerical study,we
have notpursued
this computational approach.3.5.
A
Symmetric
Case.
To
gain a better understanding of theproposed
policy,we
focuson
thesymmetric
casewhere
I=
J, a,=
a, kt=
k, 7rt=
7r, /?,•=
/?, Tj=
1, pn=
p,Ptj
—
p for i^
j (where p>
p), qn—
qand
q,j=
(I—
q)/(I—
1) for i/
j. Hence, each virus strain has thesame
parameter
values,and
for i=
1,. ..
,/, drug
combination
i is targeted at strain i. Also, let y=
Y.\=\Viand
v=
£)f=1 vt denote the totalnumber
of infected cellsand
the total free virus, respectively.
Symmetry
arguments
imply thatone
of thetwo
symmetric
static
extreme
point policies is optimal (although perhaps not uniquely so): dj(t)=
for all jand
t, or d3{t)
=
1/7 for all jand
t.Then
dt=
1—
p for all i.where p
is the "averageefficacy" of the static
drug
policy,which
is zero for the first policyand
[p+
{I—
1)p]/I forthe second policy.
The
best static policy (i.e., theone
minimizing (8)) is dj(t)=
if<
and
is dj(t)=
1/7 if>
0.where
#=
//(A-- q; airy iTii k—
oq
—
+
(<--r
Jf-)(—
r-a
A-—
a
A' A x--/'+
(v--, )-(q+
/.i)(k—
a) k-
a
k+
fi. (33)The
solution in thesymmetric
case reduces to: If 7r(1—
p)>
a
then applyno
drugs ifr-(.r-y)>—
—
-+
; (34)
~
p[?r(1
—
p)—
a\ak
if 7r(1
—
p)<
a
then applyno
treatment ifQ
+
/' ,(a
+
//)(fc+
/x) A(q+
A-+
/0r-(r-«/) <
^7—
r+
• (35)~
/3|_3r
(_1
—
p)—
aj qA'Otherwise
administerdrug combination
/*,where
v~(t)>
Uj(f) for ?=
1. ....7.Hence, the
proposed
policy in thesymmetric
case always applies the drugcombination
that corresponds to themost
prevalent virus. This therapeutic strategy certainlyseems
reasonable, although not obvious nor necessarily optimal. Also, thedrug/no drug
decision in (34)-(35)depends
onlyon
the relative value of theamount
of free virus vand
thedifferencebetween
thenumber
ofuninfectedand
infected cells. Hence, the "switching curve"(between
thedrug/no drug
decision) in three dimensions (i.e.. x, yand
v) can actuallybe
expressedas a straight line in
two
dimensions.4.
Alternative
Therapies
The
tedious part of the analysis in Section 3 is the perturbation analysis that leads tothe derivatives of the value function for a generic static policy.
Now
that these derivativeshave been estimated, it is a relatively simple matter to consider other types of therapies.
Here are two examples;
much
ofthe previous notation is reused.Protease
Inhibitors. Proteaseinhibitorsrendernewly produced
virions non-infectious.Suppose
we
haveJ
combinations of protease inhibitors,and
the controllermust
decidehow
much
of each to use subject to II,=i Tjdj(t)<
1-The
drugs' effectiveness is givenby
thematrix
p
Jnand
the resulting virus replication rateis 7rt[l
—
Y?i=\ Pjidj{t)].An
analysissimilarto that in Sectic;. 3 yields the
dynamic
indexE'"'**^*"
(36)
for drug combination j.
At
each point in time, the proposed therapeutic strategy uses the combination with the largest index if this index is positive: otherwise,no
drugs are administered.Note
that 'i^f- should be positive, so therapy should always be applied. Definedt
=
1—
J2j=iPjid*,where
dj solves the staticoptimizationproblem
that is analogous to theone in Section 3.2. Integrating the
approximate
value function inAppendix
B
with respectto Vi gives
W.
1tA
fegofe
-iW-A).
(37)dvi ki ki(ki
+
it)[p
ajkjh)
v
and
substitution of this quantity into (36) yields the proposed therapy in terms of theproblem
parametersand
the current state. For thesymmetric
case considered in Section 3.5, the solution in (36)-(37)employs
the protease inhibitor combination that correspondsto the largest value of y,{t) at each time t; that is, the therapy targets the virus strain that has infected the
most
cells.Reconstituting
the
Immune
System.
Consider using a drug, such asinterleukin-2(IL-2), that reconstitutes the
immune
system. Thisdrug
affects ourmodel
by
increasing A, the production rate of uninfectedCD4
+ cells.Suppose
thenew
production rate is A+
A(i),where
our control A(r)€
[0,A].Then
the optimizationproblem
embedded
in thedynamic
programming
optimality equation is simply tominimize A«-.
The
proposed policy is\*(t)
=
\ if^<0.
(38)ox
and
X*(t)=
otherwise,where
4^
is given in equation (28) (with dt=
1). Define theconstants
*
=
£*?-!-•
(»)
If ct
>
for all i thenwe
never use the drug,and
if c,<
for all i thenwe
always use thedrug; if neither of these cases hold then a
dynamic
policy is optimal.The
first quantityon
the right side of(39) is the expected (with respect to themutation
probabilities qtJ) value ofw/(ak). Since the
mutation
rates are very small, this quantity is nearly equal to ^/(q^A:,),and
ifwe
ignore mutations then thedrug
is always given ifand
only if 7r£
-
>
a, for all i. If~,
>
a
t. then the "basic reproductive ratio I\\," for the infected cells is greater than unity(that is, each infected cell will
produce
more
than
one
free virus particle during its lifetime),and
so thisdrug
effectively "addsmore
fuel to the fire". Empirical results (see Section 5)suggest that 7r
t
>
a,; hence,adding
uninfectedCD4
+
cells in isolation is not desirable; of
course, our
model
has not incorporated animmune
response,and
thusmay
be
omittingsome
positive side effects of additionalCD4
+ cells.Although
we
do
notdo
so here, a similar analysis canbe performed
for a therapy that reducesCD4
+ cell production.Other
therapies that canbe
analyzed include certain forms ofimmunotherapy
(whichwould
increase the death rate of free virusand/or
the death rate of infected cells), ex vivo expansion ofCD4
+ cells (Wilson et al., 1995).which would
simultaneously increase xand
decrease y;,and
dynamic
gene
therapy (Nabel, 1994),which
would
increase i\ for certainstrains.
Most
importantly,we
can also consideremploying
some
of these therapiessimultane-ously, with a joint toxicity constraint. For
example,
ifone
allowed the simidtaneous use ofRT
inhibitorsand
production ofCD4
+ cells (Schwartz et a/., 1991), itmay
turn out tobe
beneficial to introduce
CD4
+ cells at timeswhen
the infectivity of the virusand
the viral load are sufficiently suppressed.5.
An
IllustrativeExample
In this section, the
dynamic
model
is simulatedunder
theproposed
policy, as well asunder
several simplerpolicies.No
attempt
hasbeen
made
togenerate amodel
ofrealisticsize; rather,we
consider onlytwo
virus strainsand two
drug combinations in order to illustratethe nature
and
theimpact
ofdynamic
drug
treatment. In a subsequent study,we
plan touse data
from
multidrug clinical trials to analyze larger models.5.1.
Parameter
Values.
The
parameter
values for ourmodel
are displayed inTable 1.
Most
of these valueswere
sequentially derivedfrom
existing data in the followingmanner.
Ho
et al.and Wei
et al. estimate aCD4
+ production rale of roughly 1.8 x 109cells per day.
About
2%
of the totalCD4
+ population resides in the peripheral blood,and
a
human
body
contains roughly 5 x 106mm
3. Hence, the 1.8 x 109
figure is
comparable
to 7cells/mm
3 per day.The
death rate //was
chosen tobe
0.007 per day, so that the virus-freeequilibrium
CD4
+ count is A///=
1000 cells permm
3,which
corresponds to theCD4
+ count
for
an
uninfected individual.Now
we
turn to the death rates a, of infected cellsand
A*; of free virus.Ho
et al.and
Wei
et al. estimate the death rate of virus-producing cells tobe
about
0.35 per day.More
recentby. usingmore
accurate data. Perelson et al. (1995)show
that themean
death rate isabout 0.49.
They
were
also able to get arough
estimate of 3.07 for the death rate of free virus.Some
of the remainingparameters
are derivedby
considering the quasi-steady stateconditions before
drug
treatment. For typical pre-treatmentvalues,we
use the averagevalues(over 20 individuals) in Table 1 of
Ho
et a/.; the average pre-treatmentCD4
+ countwas
180cells per
mm
3and
the average viral loadwas
134 virions permm
3.Only
the wild-typevirus
was
present in nonnegligibleamounts
before treatment. Hence,we
can consider onlythe wild-type virus,
and
set the left side of equations (l)-(3) equal to zero (reflecting thequasi-steady state) to obtain a set of four equations (equations (l)-(3)
and
x+
y=
180)and
fourunknowns:
the pre-treatmentnumber
ofuninfected cells xand
infected cells y, theinfectivity rate J3
and
the replication rate K. Substituting 180-
y for x in (1)and
(2)and
solving these
two
equations for J3and
y yields J3=
(aX
—
180a/i)/(lS0m>-
\v)=
2.58x
10-4
mm
3per
day and
y=
180/)i>/(a+
fSv)=
11.86 cells permm
3. Hence, x=
168.14 cellsper
mm
3,and
the fraction of cells that are infected is y/(x+
y)=
0.066,which
is in closeagreement
with the estimate of5%
found
inEmbretson
et al. (1993). Finally, solving (3) for 7r yields k=
v(k+
0x)/y
—
35.18 virions per day, implying that tt/o=
71.8 virions areproduced
by an infected cell in our model. Readers should keep inmind
thatmost
virusis
produced
in thelymph
system,whereas
our estimates for kand $
are basedon plasma
concentrations.We
use themutation
rate calculated inMansky
and
Temin
(1995), qu
=
921=
3.4 x10"5
.
We
alsoassume
thatdrug combination
i is targeted at virus strain i.More
specifically,we
let pn
=
P22=
0.95and
p12=
P21=
0.05.meaning
that eachdrug
combination is95%
effective at blocking infections of its
own
strain, but only5%
effective at blocking infections of the other strain; such a state of affairsmight
arise if virus 1 is an AZT-resistant strain that isdominant
at time zero,and
thetwo drug
combinations correspond totwo
other (i.e., notAZT)
RT
inhibitors.The
toxicity coefficients are set to tx-
r2=
1. so that if dj(t)=
1then the
amount
ofdrug combination
j administered corresponds to the threshold toxicitylevel.
Notice that, until
now,
theparameter
values are consistent with thesymmetric
caseintroduced in Section 3.5.
Now
we
introduceasymmetry
by
letting n2=
0.9^!; hence,we
assume
that virus 1 has a higherreplication ratethan virus 2.The
pre-treatmentequilibriumstate
was
taken as the starting point of our simulation runs (see Table 1).80 100
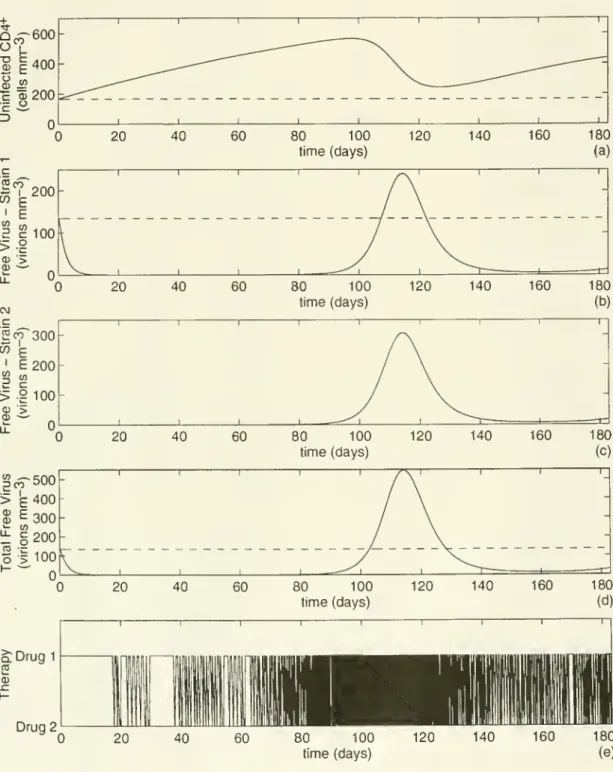
time (days)
Figure 1.
System
behaviorunder
thedynamic
policy.80 100
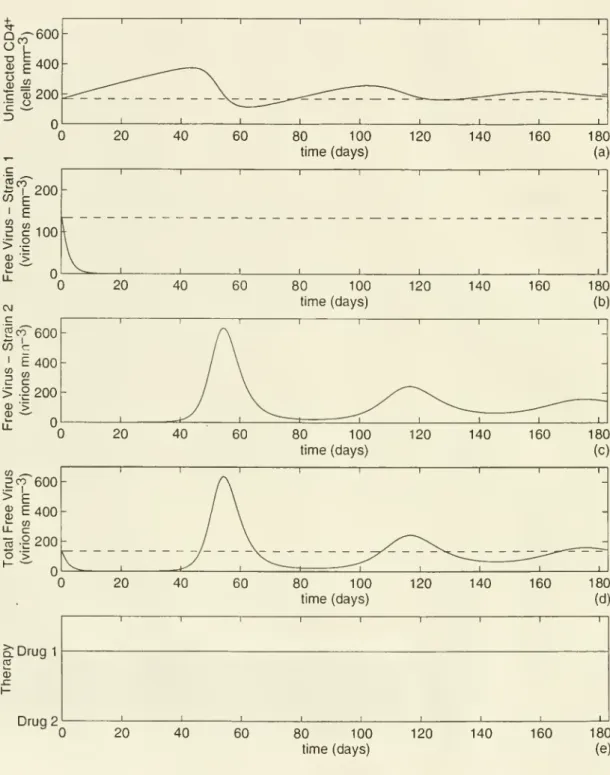
time (days)
Figure 2.
System
behaviorunder
the continuous treatment policy.the
same
quantitiesunder
the continuous treatment policy that continually applies drug 1 (i.e.. di(t)=
l.d2(t)=
for all /).Under
both
policies, theshape
of the v,and
y, curveswere very similar, with the free virus v
t lagging behind the infected cell count y,
by
severaldays; hence, the
dynamics
of the infected cellsdo
notappear
in Figures 1and
2.We
alsosimulated the
model
under
the optimal static policy derived in Section 3.2; the solutionwas
d{—
0.5-3,d
2=
0.47.Although
we
do
not include figures for the optimal static policy, theaverage (over
one
year) uninfectedCD4
+ countand
average total viral load for the variouspolicies are reported in Table 2. Readers should note that the optimal static policy
was
derivedunder
theassumption
that drug toxicities are additive; if thisassumption
is notcells,
and
this quantity peaks at threemonths;
thepeak
corresponds tomore
than a three-fold increase over the pre-treatment value.Not
surprisingly, thedynamic
policy initiatestreatment with drug 1.
The
policy first uses drug 2on
day
18,and
irregularly (e.g., it stayswith drug 1 for over a
week
at the beginning of the secondmonth)
switches backand
forthbetween
thetwo
drugsabout
once perday until theend
ofthethirdmonth. During
the fourthmonth,
thetwo
virus strains feedon
the large pool of uninfected cellsand
simultaneously emerge, peaking with a total viral load that is roughly four times the pre-treatment level.The
drug treatment oscillates rapidlybetween
thetwo
options during thismonth,
in anattempt
to simultaneously control both viruses.The
less fit virus 2 constitutesabout
55%
of the free virus during the fourth
and
fifthmonths, and hence
thedynamic
policy exerts the majority of its effort in controlling themore
fit virus 1.The
largeviral load in turn leads tomore
cell infections,and
the uninfectedCD4
+ countdecreases in the fourth
month, bottoming
out at a level that is higher than thepre-treatment uninfectedCD4
+ count. This reduction in uninfected cells allows the drugs to bringboth
viruses back
under
control during the fifthmonth.
After this time, theseoscillationsdampen
(e.g., the total viral load peaks again at theend
ofthe seventhmonth
with a value ofabout
240)and
anew
steady state is slowly reached.Over
the first sixmonths,
thedynamic
policy allocated58.7%
of the time to drug 1and 41.3%
of the time todrug
2: hence, thedynamic
policy
expended
more
effort than the optimal static policyon
themore
fit virus.Continuous
Treatment.Under
the continuous treatment policy pictured in Figure 2,we
again see arapiddrop
in virus 1and
alinear risein theCD4
+ count; however, in contrastto the
dynamic
policy, the continuous treatment policy suppresses virus 1 throughout thefirst six
months.
Virus 2 emerges during the secondmonth,
and
reaches a very high level bythe
end
of themonth.
This high viral load then drops the uninfectedCD4
+ countbelow
itspre-treatment level,
which
in turn leads to a reduction of free virus 2 in the thirdmonth.
Comparison
ofPolicies. Table 2shows
that thedynamic
policy performsmuch
betterthan the continuous treatment policy: the continuous policy achieves a
17.4%
reduction in average (over the first sixmonths)
viral loadand
a31.6%
increase in uninfectedCD4
+ count with respect to the drug-free equilibrium, whereas thedynamic
policy achieves a53.4%
reduction in viral loadand 126.2%
increase in uninfectedCD4
+ count. Moreover, the viral peaksand
CD4
+ valleys are lesspronounced under
thedynamic
policy thanunder
thecontinuous treatment policy. Finally, the
dynamic
policy, by frequently switchingbetween
thetwo drug
options during the earlymonths,
delays theemergence
of virus 2from
thesecond
month
until the fourthmonth.
Although
the results are not reported here,we
alsotested
an
alternating policy that usesdrug
1 for the first threemonths
and
then uses drug2 for the last three
months. Not
surprisingly, this policy did notperform
well.We
should note that the post-treatment steady state values of uninfectedCD4
+ cellsand
total virus loadunder
all of the therapeutic strategies are not appreciably differentthan the pre-treatment steady state values; hence, the therapeutic benefits in our
model
are achieved during the transientdomain.
However,
the viraldynamics
of HIV-infectedindividuals
undergoing
dynamic
drug treatmentwould
probably exhibit transient behavior formany
years. Therefore, theimprovements
over the transientdomain
in ourmodel
shouldbe
indicative of theimprovements
that canbe
realized in clinical practice.Unequal
Infectivity Hates.Because
thereseems
tobe
some
uncertaintyabout
whether
virus strains have different infectivity rates,
we
reduced the infectivity of virus 2 so that 02=
0.9/?i-The
qualitative resultswere
similar to our base case, although thedynamic
policyexpended
a slightly larger fraction of its efforton
virus 1, resulting in asomewhat
larger proportion of virus 2 in the viral mix.
More
generally,numerous
other simulationruns
were
generatedby
varying theparameters
7r£-,$,
Aand p
(J,and
the qualitative resultsdisplayed in Figures 1
and
2remained
intact; hence, themodel
appears tobe
robust.7.
Conclusions
We
have used the control theoryparadigm
in aHIV
therapeutic setting.Our
model
incorporates different virus strains,and
a variety of therapeutic options are available.The
approximation
method, which
uses perturbation analysisand
the policyimprovement
al-gorithm, gives rise to a
dynamic
index policy: eachdrug
combination has an associateddynamic
index,and
at each point in time the policy uses the drug combination with thelargest index.
The dynamic
indices succinctlysummarize
three quantities: the efficacy ofeach drug combination
on
each virus strain, the toxicity ofeachdrug
combination,and
themarginal benefit of blocking a
new
cell infectionby
each virus strain; the last ofthese three quantities changes over time as a function of an individual'sCD4
+ count, viral loadand
viral mix.Numericalresults for atwo-virus,two-drug
model
suggest thatdynamic
multidrug ther-apiesoutperform
their static counterparts: the total viral load is reduced, the uninfectedCD4
+ count is increased,and
theemergence
of drug resistant strains is delayed.Although
the individualized therapy thatwe
propose isno doubt
more
difficult toimplement than
protocols that are currently in practice, the benefitsmay
outweigh
the costs ofimplemen-tation.
These
benefits are achievedby
frequently changing therapies over time in responseto
and
in anticipation oftheemergence
of drug-resistant strains. In addition, thedynamic
policy attempts to maintain themore
fit virus strains at a relatively low level, while perhapsallowing the less fit strains to partially establish themselves.
Although
our numerical results focuson
the two-virus, two-drug case, thedevelopment
of
more
realisticmodels
using datafrom
multidrug studies isplanned
for the future. Itmay
turn out that the best
way
to delay the onset ofAIDS
is via the intelligent use of awide
range of therapies(RT
inhibitors, protease inhibitors, reconstitution of theimmune
system,immunotherapy,
gene therapy, etc.).The
model and
analysis presented here provides theframework
for thedevelopment
of such therapeutic strategies.Finally, our
approach
to thisproblem
circumvents the usual obstacles inherent insolv-ing high-dimensional nonlinear control problems. This
method, which
appears to be new, has potential applications in awide
variety of controlproblems
in epidemiologyand
ecology: besides allowing formutation
among
multiple variants of entities (in this case, viruses), theapproach
can also incorporate discrete age classes (e.g., for optimal vaccinations of measles)and
discrete spatial (e.g.. lattice) structures (e.g., fordynamic
control of spatial epidemics).Acknowledgment
The
first authorwas
supportedby
NSF
grantDDM-9057297
and
EPSRC
grantGR/J71786.
He
gratefully acknowledges a valuable discussion with Peter Whittle during the early course ol this research,and
thanks Denise Kirschner for a helpful conversationabout parameter
estimation.The
second authorwas
supportedby
theWellcome
Trustand Keble
College.REFERENCES
Agur. Z. (1989).
A
new method
for reducing cytotoxicity of theanti-AIDS
drugAZT.
Biomedical
Modeling
and
Simulation. Levine, D. S., ed., J. C. BaltzerAG,
ScientificPub-lishing Co.
IMACS.
59-61.Avriel,
M.
(1976). NonlinearProgramming.
Prentice-Hall,Englewood
Cliffs, NJ. Bellman, R. E. (1957).Dynamic
Programming.
Princeton U. Press, Princeton, NJ.Condra. J. H. et al. (1995). In vivo
emergence
ofHIV-1
variants resistant to multiple protease inhibitors.Nature
374, 569-571.Elliott, R. J.,
Aggoun,
L.and Moore,
J. B. (1994).Hidden
Markov
Models: Estimationand
Control. Springer- Verlag.New
York.Embretson.
J. ef al. (1993). Analysis ofhuman
immunodeficiency
virus-infected tissuesby
amplificationand
in situ hybridization reveals latentand
permissive infections at single-cellresolution. Pvoc. Natl. Acad. Sci.
USA
90, 357-361.Frost, S. D.
W.
and
McLean,
A. R. (1994). Quasispeciesdynamics and
theemergence
ofdrug resistance during zidovudine therapy of
HIV
infection.AIDS
8. 323-332.Gittins, J. C. (1989).
Multi-armed
Bandit Allocation Indices. Wiley,New
York.Golub. G. H.
and
Van
Loan, C. F. (1989).Matrix Computations. Johns Hopkins
U. Press,Baltimore.
Ho. D. D. et al. (1995).
Rapid
turnover ofplasma
virionsand
CD4
lymphocytes
inHIV-1
infection.
Nature
373, 123-127.Howard,
R. A. (1960).Dynamic
Programming
and
Markov
Processes.MIT
Press,Cam-bridge,
MA.
Key. P. B. (1990).
Optimal
controland
trunk reservation in loss networks. Prob. Engrg.Inf. Sci. 4. 203-242.
Kirschner. D., Lenhart. S.
and
Serbin. S. (1995).Optimal
control of thechemotherapy
ofHIV.
Preprint, Dept. of Math., U. of Tennessee, Knoxville,TN.
Kirschner. D.
and
Perelson. A. (1994).A
model
for theimmune
system
response toHIV:
AZT
treatment studies.Mathematical
PopulationsDynamics
III,Theory
of Epidemics.Arino. 0.. Axelrod, D.
and Kimmcl.
M.. eds., 1.Wuerz
Publ..Winn.
Manitoba.Kirschner, D.
and
Webb.
G. F. (1995a).A
model
for treatment strategy in thechemotherapy
of
AIDS. Submitted
to Bull.Math.
Biology.Kirschner. D.
and
Webb.
G. F. (1995b). Effects ofdrug
resistanceinchemotherapy
strategiesin the treatmentof
HIV
infection. Preprint, Dept. of Math..Texas
A&M
U.. College Station.TX.
Kojima.
E. ef al. (1995).Human
immunodeficiency
virus type 1 (HIV-1) viremia changesand development
of drug- relatedmutations
in patients withsymptomatic HIV-1
infectionreceiving alternating or simultaneous zidovudine
and
didanosine therapy. J. Infectious Dis-eases 171. 1152-1158.Lagakos. S.. Pettinelli.
C.
Stein. D.and
Volberding. P. A. (1993).The
Concorde
Trial. Lancet 341. 1276.Larder. B. A.. Kellam. P.
and
S. D.Kemp.
(1993).Convergent
combination therapy can select viablemultidrug-resistantHIV-1
in vitro.Nature
365. 151-453.Larder. B. A..
Kemp.
S. D.and
Harrigan. P. R. (1995). Potentialmechanism
for sustainedantiretroviral efficacy of
AZT-3TC
combination
therapy. Science 269. 696-699.Mansky.
L.M. and Temin,
H.M.
(1995).Lower
in vivomutation
rate ofhuman
immunod-eficiency virus type 1 than that predictedfrom
the fidelity of purified reverse transcriptase.J. Virology 69, 5087-5094.
McLean.
A. R.and
Nowak. M.
A. (1992).Competition between
zidovudine sensitiveand
zidovudine resistant strains of
HIV.
AIDS
6. 71-79.McLeod.
G. X.and
Hammer.
S.M.
(1992). Zidovudine: 5 years later.Annals
of InternalMedicine
117. 487-501.Nabel. G. J. (1994).
Gene
therapy approaches toAIDS.
AIDS
8. S61-S69.Nowak.
M.
A. et al. (1991). Antigenic diversity thresholdsand
thedevelopment
ofAIDS.
Science 254. 963-969.
Nowak. M.
A.and May.
R.M.
(1993).AIDS
pathogenesis:mathematical models
ofHIV
and
SIV
infections.AIDS
7. S3-S18.Ott. T. J.
and Krishnan
K. R. (19S5). Statedependent
routeing of telephone trafficand
theuse of separable routeing schemes. In Proc. 11th Int. TeletrafRc Cong..
Akiyama,
M.. ed..Elsevier,
Amsterdam.
Perelson, A. S. (19S9).
Modeling
the interaction of theimmune
system
withHIV.
In Mathe-maticaland
StatisticalApproaches
toAIDS
Epidemiology, Castillo-Chavez,C,
ed., Lecture Notes in Biomath., Springer- Verlag,New
York, 83, 350-370.Perelson, A. S., Kirschner, D.
and DeBoer,
R. (1993).The
dynamics
ofHIV
infection ofCD4+
cells.Math.
Biosciences 114, 81-125.Perelson, A. S. ef al. (1995).
HIV-1
dynamics
in vivo: virion clearance rate, infected cell lifespan,and
viral generation time.Submitted
to Science.Pontryagin, L. S., Boltyanskii, V. G., Gamkrelidze, R. V.,
and
Mishchenko, E. F. (1962).The
Mathematical
Theory
ofOptimal
Processes. Interscience Publishers,New
York.Richman,
D. D. et al. (1994). Resistance toAZT
and
ddC
during long-term combination therapy in patients withadvanced
infection withhuman
immunodeficiency
virus. JAIDS
7,135-138.
St. Clair.
M.
II. ef a/. (1991). Resistance to ddland
sensitivity toAZT
induced by amutation
inHIV-1
reverse transcriptase. Science 253, 1557-1559.Schuurman,
R. et al. (1995).Rapid
changes inhuman
immunodeficiency
virus type 1RNA
load
and appearance
of drug-resistant virus populations in persons treated with lamivudine (3TC). J. Infectious Diseases 171. 1411-1419.Schwartz, D. H.,
Skowron,
G.and
Merigan, T. C. (1991). Safetyand
effects of interleukin-2 plus zidovudine inasymptomatic
individuals infected withhuman
immunodeficiency
virus.J
AIDS
4. 11-23.Shafer. R.
W.
ef al. (1991).Combination
therapy with zidovudineand
didanosine selectsfor drug-resistant
human
immunodeficiency
virus type 1 strains with unique patterns of polgene mutations. J. Infectious Diseases 169. 722-729.
Shafer, R.
W.
et al. (1995).Drug
resistanceand
heterogeneous long-term virologicresponsesof
human
immunodeficiency
virus type Tinfected subjects to zidovudineand
didanosinecombination therapy. J. Infectious Diseases 172. 70-78.
Shaw,
G.M.
(1995). Personalcommunication.
Volberding, P. A. ef al. (1994).
The
duration of zidovudine benefit in persons with asymp-tomaticHIV
infection.JAMA
272, 437-442.Wei, X. et al. (1995). Viral
dynamics
inhuman
immunodeficiency
virus type 1 infection.Nature
373, 117-123.Wilson, C. C.
and
Hirsch.M.
S. (1995).Combination
antiretroviral therapy forthe treatmentof
human
immunodeficiency
virus type-1 infection. Proc. Ass.American
Physicians 107.19-27.
Wilson. C. C. et al. (1995).
Ex
vivo expansion of CD-Ilymphocytes
from
human
immunod-eficiency virus type 1-infected persons in the presenceof
combination
antiretroviral therapy.J. Infectious Diseases 172. 88-96.
APPENDIX
A
The
solution to (24)- (27) isi=i
H
\(ki-
oti)([i-
cti) ki-otin-ki
+
ft*(*
-
-)
^77
r-
+
&
-
-^-)
. (40)/' \ cti(ki-ai) A'i-a, A-,
JJ
// \ A',
—
Q
t fcj—
Q
; a,—
A'qti&idt(x
—
)(i
^/ \ ^(fcj
-
a,) A-,-
a, a,-
A-,-
///+
fc^-d, *-
-)
j7j+
{Vj-
—
1±-)
. (41 //\{kj-
ocj){Cti-
a
j-
fi ) kj-
c\j o t-kj.-fiJJ
.. A / iriyie-l°i-+ M-t) . 7r ltj t c -(*+rt(-t)\-Pi(x
-
-)
ji -rr, r-
(»<-
j )/' \{ki
-
ai){ki-
cti-
n) ki-
a, // /fi \
{ki-ctiy
ki-ai
ki-cti aii-ki J\ ( ir-?/f-(ai+")(s-') it a P-(fc.+/x)(s-t) \
-maPidiix
~
-)
-77 rr; r+
(r<_
1 )' /' \f'{k,-
a,){k,-
a,•-
//) fc;-
a; //(a,-
A-(-
//) /+
^r'"
Ji 'V
Uibj-
*,-)(<*-
a
j)(kt-
a,-)+
(r '"
*,•-Vte
-
*,-)(**"
^)J
+
Xiqjiftdjix—)
—
^
+
(v 3-
T
jMl
-h
:m
7 (42)where
the constants appearing in these equations arec,=i(-T^-.L --**-)-%* --i(^
+
.-.))(«)
^y,
=
-QiiPidi- ( 7—
+
{Vi-[t VA'j
—
Q, A,—
Q; q,—
Av+
9iiftdI- j--
-)(-7]
--('-,-i ) ; -)
A
*iVi
(aj
-
%)(«<
-
a,)kj-aj
(ai-kj
+
qJl3
JdJ{x *iVj fi \{kj-a
}){Qj +fi-a
tC
VlTil _ A / 7T;t/j-
to
-
T
2
*-)"
kj-
a
} q,-
kj-
fi] KiVia,
=
~^±-
+
ft-
IT^^TT
+
(«i-
^-)<
+ft(x-fcj-
a, "'// V(^«_Q
<) 2 A /^
ft \(ki
—
ati)(ki—
cti—
p) (Vi -TTiqufiidi- ( ,, ' [„
(*-
-,-)
+
(vik
-
an ft \(ki-
a
t) 2 ki-
a, A+
7T tqit3ldt{x ) n-«y,+
H
("-'/,..V'j
// \//(A*,-
Cti)(ki-
Q,-
//) j/<fa
A -,—
Q, Q,—
A'ly fcj-
Q
( /((a,-
A,-
fi) (44);Aj
-
a,)(o,-
a,-)(fci_
Q
j) fc j_
a
j (a
«~
fcj)(^~
fci; A Xiqjifydjix ) 77,11,+
(»i-7T, 2/j ): l// \(&j
-
Qj)(Qi;-Qj
-
/<)(*";-
«j-
/0^i-«j
(Ot-^i
-/*)(*^
-
a':(45)
APPENDIX
B
The
approximate
value functionV
e is givenby
rT I
f
ECt^OO
+
^OO)*
L
77 ;—
+
(u«"
7 )_
fc+
eC
w
*«(l-
e-'tr-*>)C„(i-
e-*iCr-t))0,(^
-
"t;+
ftA/^^e-^
1"-*)-!: ( // va
t(&i-
a
t) 2+
(Vi «iVih
-
a,- 1,2 ) A+Pi{x-
-)
TTiJ/i ( -i >,+u)(T-t)_ y
+
(», - //, 1-
c-(^+"Hr-n
//' MA', -Q,)(I',
-
Qj-
fi){al;+
fi)ki-Oi
Vih
+
V)