Dimensionality Reduction in immunology: from
viruses to cells
by
Karthik Shekhar
B. Tech. in Chemical Engineering
M. Tech. in Chemical Engineering
Indian Institute of Technology Bombay (2008)
Submitted to the Department of Chemical Engineering in partial
fulfillment of the requirements for the degree of
Doctor of Philosophy
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
@2014
Massachusetts Institute of Technology. All rights reserved.
Author ...
Certified by.
July 2014
[lf e
Dr.oam
2
oi5
3ignature
redacted
S
Department of Chemical Engineering
July 14, 2014
ignature redacted
Arup K. Chakraborty
Robert T. Haslam Professor of Chemical Engineering
Thesis Supervisor
Accepted by..
Signature redacted
...
Patrick S. Doyle
Professor of Chemical Engineering
MASSACHUSETTS INSTITUTE OF TECHNOLOLGY
JUN 16 2015
This doctoral thesis has been examined by a Committee of the
Department of Chemical Engineering as follows:
Professor
J.
Christopher Love...
...
Latham Family Career Development Professor of Chemical
Engineering
Committee Chair
Professor Arup K. Chakraborty ...
Director, Institute for Medical Engineering and Science
Robert T. Haslam Professor of Chemical Engineering
Professor of Physics, Chemistry and Biological Engineering
Thesis Supervisor
Professor M ehran Kardar...
Francis Friedman Professor of Physics
Committee Member
Dr. Bruce D . W alker, M D ...
Director, Ragon Institute of MGH, MIT and Harvard
Abstract
Developing successful prophylactic and therapeutic strategies against infections of RNA viruses like HIV requires a combined understanding of the evolutionary constraints of the virus, as well as of the immunologic determinants associated with effective viremic control. Recent technologies enable viral and immune pa-rameters to be measured at an unprecedented scale and resolution across multi-ple patients, and the resulting data could be harnessed towards these goals. Such datasets typically involve a large number of parameters; the goal of analysis is to infer underlying biological relationships that connect these parameters by ex-amining the data. This dissertation combines principles and techniques from the physical and the computational sciences to "reduce the dimensionality" of such data in order to reveal novel biological relationships of relevance to vaccination and therapeutic strategies. Much of our work is concerned with HIV.
1. How can collective evolutionary constraints be inferred from viral sequences derived from infected patients? Using principles of Random Matrix Theory, we derive a low dimensional representation of HIV proteins based on circu-lating sequence data and identify independent groups of residues within vi-ral proteins that are coordinately linked. One such group of residues within the polyprotein Gag exhibits statistical signatures indicative of strong con-straints that limit the viability of a higher proportion of strains bearing multiple mutations in this group. We validate these predictions from
in-dependent experimental data, and based on our results, propose candidate immunogens for the Caucasian American population that target these vul-nerabilities.
2. To what extent do mutational patterns observed in circulating viral strains accurately reflect intrinsic fitness constraints of viral proteins? Each strain is the result of evolution against an immune background, which is highly diverse across patients. Spin models constructed to reproduce the preva-lence of sequences have tested positively against intrinsic fitness assays (where immune selection is absent). Why "prevalence" should correlate with "replicative fitness" in the case of such complex evolutionary dynam-ics is conceptually puzzling. We combine computer simulations and ana-lytical theory to show that the prevalence can correctly reflect the fitness rank order of mutant viral strains that are proximal in sequence space. Our
analysis suggests that incorporating a "phylogenetic correction" in the pa-rameters might improve the predictive power of these models.
3. Can cellular phenotypes be discovered in an unbiased way from high di-mensional protein expression data in single cells? Mass cytometry, where
> 40 protein parameters can be quantitated in single cells affords a route, but analyzing such high dimensional data can be challenging. Traditional "gating approaches" are unscalable, and computational methods that ac-count for multivariate relationships among different proteins are needed. High-dimensional clustering and principal component analysis, two ap-proaches that have been explored so far, suffer from important limitations. We propose a computational tool rooted in nonlinear dimensionality reduc-tion which overcomes these limitareduc-tions, and automatically identifies pheno-types based on a two-dimensional distillation of the cellular data; the latter feature facilitates unbiased visualization of high dimensional relationships. Our tool reveals a previously unappreciated phenotypic complexity within murine CD8+ T cells, and identifies a novel phenotype that is conflated by traditional approaches.
4. Antigen-specific immune cells that mediate efficacious antiviral responses in infections like HIV involve complex phenotypes and typically constitute a small fraction of the population. In such circumstances, seeking correl-ative features in bulk expression levels of key proteins can be misleading. Using the approach introduced in 3., we analyze multiparameter flow cy-tometry data of CD4+ T-cell samples from 20 patients representing diverse clinical groups, and identify cellular phenotypes whose proportion in pa-tients is strongly correlated with quantitative clinical parameters. Many of these correlations are inconsistent with bulk signals. Furthermore, a number of correlative phenotypes are characterized by the expression of multiple proteins at individually modest levels; such subsets are likely be missed by conventional gating strategies. Using the in-patient proportions of different phenotypes as predictors, a cross-validated, sparse linear re-gression model explains 87 % of the variance in the viral load across the twenty patients. Our approach is scalable to datasets involving dozens of parameters.
Acknowledgments
To the following, this dissertation and I owe an inexpressible measure of grati-tude.
To Arup Chakraborty, for his unbridled enthusiasm of science, generosity and wisdom. To Profs. Bruce Walker, Mehran Kardar and Chris Love for their support and enthusiasm of my work. To Vincent Dahirel for his mentorship and support during my teething days in graduate school.
To Petter Brodin for providing me a much-needed intellectual break, and for his continuing support and collaboration. To Prof. Mark M. Davis for his encour-agement and support for our project when Petter and I needed them. To Damien Soghoian for another enthusiastic collaboration. To former and current members of the Chakraborty group (particularly Vincent, Steve, Andy, Tom, Misha, Ming and Shenshen) for all the camaraderie and moral support.
To Don and Elizabeth without whose tireless efficiency my life as a graduate student (and those of many others in the group) would be far more difficult. To Suzanne and Joel from the MIT ChemE students office, for all their indispensable support. To the Jerry and Geraldine S Mcafee fellowship, the Poitras pre-doctoral fellowship and the Ragon Institute for financial support. To MIT, where the stars
are closest to the earth for young scientists like me.
To my friends from IIT Bombay -Manas, Niranjan, Kaushik, Onkar, Parasvil, Purushottam, Sarmistha, Sudeep, Sumedh, Sushant, Varun, Vignesh -the warmth, wit and humor you radiate has not been diluted by distance. To the wonderful friends I made in this country - Aravind, Dipti, Diwakar, Keerthana, Kavita, Leonid, Neela, Nitin, Paul, Pavan, Pooja, Premnandhini, Priya, Shreya, Umang
-to whom my life during the past five years owes uncountable favors, small and big. To Jonathan and Somnath, whose selfless concern for humanity and suffer-ing in this world despite every personal challenge, continues to be an inspiration. To my parents Shekhar and Vijaya, both my work and my person owe great debt. They have encouraged and supported my aspirations, irrespective of what it meant for their comfort. My failures are my own and my successes, I owe to their nurturing.
To the rest of my family, for their unstinted support and enthusiasm for my intellectual pursuits. I am particularly grateful to my grandparents (paternal and maternal), my maternal aunt Bhavani, my sister Nandini, cousin Raja. To my now expanded family - my parents in-law and my brother in-law Varun - for their faith in me and my abilities.
To my wonderful wife Veena, whose warmth and compassion know no bounds; this thesis owes as much to her nurturing as it does to my labor. To our darling daughter Veda, who unknowingly brings much peace and happiness to her fa-ther's perennially restless mind.
Contents
1 Introduction 17
1.1 The Adaptive Immune System - A primer ... 22
1.1.1 B cells and T cells - different arms of adaptive immunity . . 22
1.1.2 Different modes of recognition of pathogens and immuno-logical m em ory . . . . 23
1.2 Collective evolutionary constraints in HIV proteins and its rele-vance for rational immunogen design . . . . 25
1.2.1 In vivo characteristics of HIV infection . . . . 26
1.2.2 Implications of HIV mutability for therapies and vaccines 28 1.2.3 Importance of CD8+ T-cell responses against HIV, and im-plications for a T-cell vaccine . . . . 30
1.2.4 The immunogen design question . . . . 34
1.3 Faithful inference of intrinsic fitness landscapes from patient-derived viral sequence data . . . . 36
1.3.1 What are viral fitness landscapes? . . . . 36
1.3.2 Inferring fitness landscapes from data . . . . 37
1.3.3 Agreement with RC measurements and the puzzle . . . . . 39
1.4 Identification of cellular phenotypes from high dimensional ex-pression data . . . . 41
1.4.1 Measuring single-cell phenotypes . . . . 41
1.4.2 Computational challenges involving the analysis of high di-mensional data . . . . 42
1.5 Identification of CD4+ T-cell phenotypes correlated with HIV con-tro l . . . . 45
1.5.1 The "quality" of the T-cell response . . . . 45
1.5.2 The role of quality in mediating viral control . . . . 46
1.6 Dimensionality Reduction . . . . 49
1.7 Statement of Collaborations . . . . 50
2 Coordinate linkage of HIV evolution reveals regions of immunologic vulnerability 53 2.1 Introduction . . . . 54
2.1.1 Analogy with Stock Market Analysis . . . . 56
2.2 R esults . . . . 58
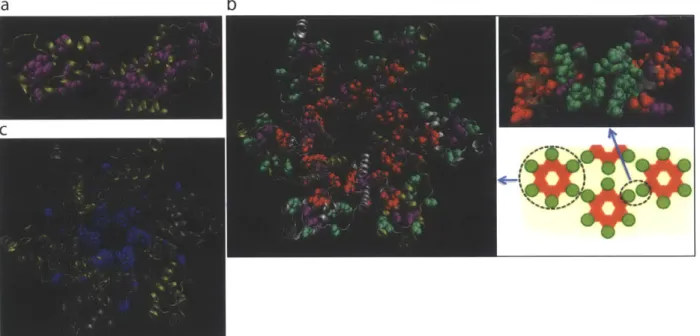
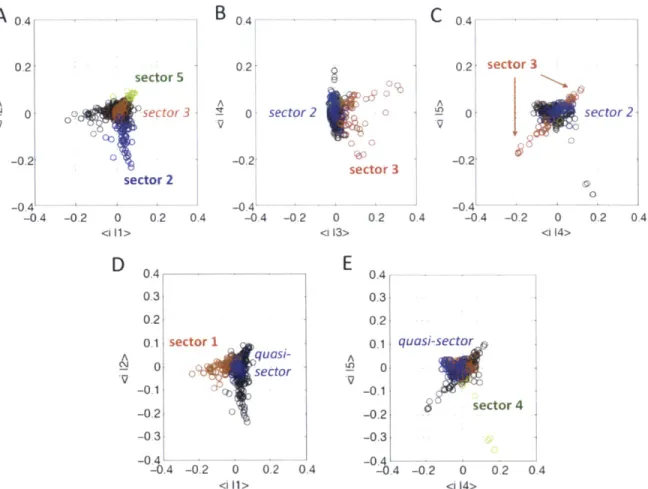
2.2.1 RMT analysis and identification of HIV Gag sectors ... 59
2.2.2 An immunologically vulnerable sector in Gag . . . . 65
2.2.3 Sector 3 is present at critical oligomeric interfaces of the viral cap sid . . . . 68
2.2.4 Elite controllers of HIV preferentially target multiple sites in sector 3 . . . . 70
2.2.5 Virus sequences from patients show that multiple mutations in sector 3 are rare . . . . 73
2.2.6 Immunogens that may induce CTL responses in a popula-tion that hurt HIV . . . . 75
2A-1 Definition of the correlation matrix . . . . 78
2A-2 The spectral contribution from phylogeny - A pedagogical model 80 2A-3 Removing the contribution of phylogeny from the correlation matrix 82 2A-4 Removing evolutionarily distinct sequences . . . . 85
2A-5 Defining sectors . . . . 87
2A-6 Determining Gag sectors . . . . 88
2A-7 A quasi-sector induced by immune pressure . . . . 90
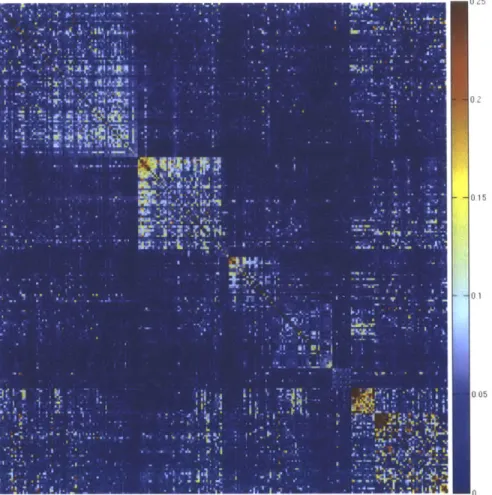
2A-8 Representation of the cleaned correlation matrix . . . . 92
2A-9 Association between immune pressure in controllers and sectors . 94 2A-10 Strength of negative correlations . . . . 99
2A-11 Immunogen design . . . . 102
2A -12 N ef sectors . . . 106
2A-13 Reverse Transcriptase (RT) sectors . . . 109
2A-14 Distribution of significant positive and negative correlations using different thresholds . . . .111
2A-15 Validation of predictions with available data and new predictions 114 2A-16 Evaluation of three-site correlations . . . 116
2A-17 Analysis of Sequences from Elite Controllers . . . 116
2A-18 Ragon collaborative study on sector immunogens . . . 117
2A-18.1 Computational identification of immunogens using fitness landscapes ... ... 119
2A-18.2 Additional considerations related to fitness cost . . . . 123
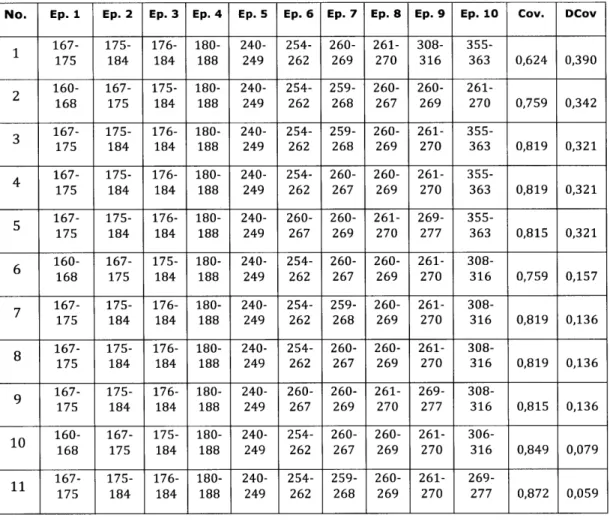
2A-18.3 Human Epitopes within included chunks . . . 123
3 Spin models inferred from patient-derived viral sequence data
3.1 Introduction . . . . 3.1.1 Connecting prevalence and intrinsic fitness . . . . 3.2 Simulations . . . . 3.3 R esu lts . . . . 3.4 Variational theory . . . . 3.5 Discussion . . . . 3A-1 Modeling immune pressure . . . . 3A-1.1 Statistics of T cell targeting of p17 . . . . 3A-2 Quasispecies simulations . . . . 3A-3 Sampling Hit
[s]
and low-dimensional representation using PCA 3A-4 Landscape visualization . . . . 3A-5 Comparison of mutational probabilities upon restricted sampling 3A-6 Robustness studies . . . .3A-6.1 The mutation rate y . . ...
3A-6.2 Gaussian distributed {bi} 3A-6.3 Uniformly distributed
{bi}.
3A-6.4 Number of generations . . . 3A-6.5 Changing nmax . . . .3A-7 Variational calculations . . . .
. . . 1 6 1 . . . 16 4 . . . 16 6 . . . 16 7 . . . 16 8 . . . 1 7 1
4 Automatic cell classification from high-dimensional mass data
4.1 Introduction . . . . 4.1.1 Technologies to interrogate single cells . . . . 4.1.2 Data analysis . . . . 4.2 R esults . . . . cytometry 177 178 178 179 183 126 128 130 134 138 144 146 148 149 153 156 159 159
4.2.1 4.2.2 4.2.3 4.2.4 4.2.5 Computational Methods ...
Analyzing CD8+ T-cell populations in SPF mice using t-SNE Phenotypic coarse-graining ...
Analysis of identified CD8+ T-cell subpopulations in mice Comparison of CD8 T-cell subpopulations between specific pathogen free (SPF) and germ-free mice . . . .
183 185 187 188 192 4.3 D iscussion . . . 196
4A-1 Data collection and preprocessing . . . 198
4A-1.1 Mice used in the experiments . . . 199
4A-1.2 Staining panel . . . 200
4A-1.3 Data generation, instrument settings, data transformation and data normalization . . . 200
4A-1.4 Batch effects . . . 202
4A-1.5 Density dependent down sampling . . . 203
4A-2 Mathematical overview of t-SNE . . . 204
4A-2.1 A note on the dimensionality of the embedding . . . 206
4A-2.2 Avoiding dimensionality reduction altogether . . . 207
4A-3 Numerical procedure to compute the embeddings . . . 208
4A-4 Identification of subpopulations . . . 209
4A-5 Projecting additional points in the t-SNE map . . . 212
4A-6 Principal Component Analysis (PCA) . . . 215
4A-7 Protein expression in different subsets . . . 218
4A-7.1 Phenotypic signatures of SPF B6 subsets . . . 220 4A-7.2 Phenotypic signatures of T-cell subsets S1 - S8 in GF mice 240
5 Correlative features HIV control in antigen-specific CD4+ T cells from 12-color flow cytometry
5.1 Introduction . . . . 5.2 Dimensionality reduction using ACCENSE . . . . 5.2.1 Exploring the low dimensional representation . . . . 5.3 R esu lts . . . . 5.3.1 Correlational analysis of bulk protein expression . . . . 5.3.2 Analysis of multivariate expression . . . . 5.3.3 Correlations between clinical status and proportions of phe-notypic subpopulations in patients . . . . 5.4 Correlations between clinical indicators and proportions of
phe-notypic subpopulations in pati( 5.5 Multivariate expression signatu 5.6 Reconciling bulk expression lex 5.6.1 Granzyme B . . . . 5.6.2 IL-2 . . . . 5.7 A multiple regression analysis 5.8 Summary . . . . 5A-1 Experimental Methods . .
5A-1.1 Stimulation of cells .
5A-1.2 Flow cytometric staining 5A-1.3 Flow cytometric analysis 5A-2 Additional figures and tables .
n ts . . . 2 6 7 ires . . . 2 69 rels . . . 2 74 . . . 2 74 . . . 2 75 . . . 2 76 . . . 2 77 . . . 2 79 . . . 2 8 1 . . . 2 8 1 . . . 2 82 . . . 2 82
6 Conclusions and Outlook 291
247 248 249 250 252 252 255 258
6.1 Evolutionary constraints of viral proteins towards rational design of immunogens . . . 293 6.2 Identification of multivariate phenotypes from high dimensional
1
Introduction
Biological systems typically feature a large number of components interacting in complex ways at different spatiotemporal scales. The vertebrate immune system, which protects hosts from opportunistic pathogens through a variety of defense mechanisms, is no exception to this rule. Recent surges in technology have en-abled experimentalists to interrogate the immune system at various levels from the molecular to the cellular and the systemic at an unprecedented resolution and throughput [1, 21. The resulting data provide opportunities to infer the mechanistic and organizational principles underlying the immune system, and to develop targeted interventional strategies that can improve human health in dis-eased states. This thesis seeks to make a small advance by combining techniques from the physical and the computational sciences towards such "data-driven in-ference" in immunology. Since such data typically involve large numbers of vari-ables, we employ approaches that seek to "reduce the dimensionality" towards revealing the most useful features, and sieving away those that are irrelevant.
The adaptive immune system in jawed vertebrates refers to a collective of
cell-types (e.g. B and T cells), organs (e.g. thymus, lymph nodes, spleen), and various processes that recruit these components (e.g. inflammation, affinity maturation,
antigen presentation) to protect the host from opportunistic pathogens like bac-teria, viruses, parasites and fungi [3]. The ability of the adaptive immune system to mount responses that are highly specific to the infectious agent despite the absence of ex ante genetic preprogramming makes it distinct from the innate
im-mune system [4]. Furthermore, the adaptive immune system "learns" from every infection, which enables it to rapidly respond upon subsequent invasions by the same pathogen - this remarkable capacity, termed "immunological memory",
forms the basis of the modern science of vaccination [5]. In what follows we review some of these concepts and mechanisms briefly. The reader may refer to standard textbooks on immunology for greater depth [3, 61.
RNA viruses are the causative agents for numerous infectious diseases that affect animal and human life around the world -e.g. influenza, Foot and Mouth Disease Virus (FMDV), Hepatitis C Virus (HCV) and Human Immunodeficiency Virus (HIV) [7]. A great degree of public health and scientific effort continues to be focused on combating these diseases - these range from measures to im-prove sanitation, public outreach, innovation in vaccines and drugs. Owing to a number of common biological factors (e.g. small genome sizes, error-prone replication, high fecundity, the ability to exploit sociological characteristics of the host, and the capacity to manipulate host physiology to their own ends) and other specific ones (e.g. impairing immune cell function (HIV), genetic reas-sortment (influenza)), these viruses continue to adversely impact human health despite interventions against them [8]. They evade vaccine-induced immune re-sponses and become drug-resistant, adapting and persisting against our efforts to eradicate them.
muta-tion is a serious barrier to the design of effective prophylactic and therapeutic strategies. For e.g., the design of an influenza vaccine is a yearly exercise that entails a considerable degree of predictive uncertainty (not to mention looming threat of a pandemic outbreak every now and then); in contrast, the prospects for an efficacious HIV vaccine remain uncertain despite years of efforts and bil-lions of dollars. HIV is unprecedented in its capacity to mutate and diversify [9]. A departure from conventional vaccination strategies has become imperative to confront this challenge. Information regarding the evolutionary constraints of HIV at the molecular detail can inspire rational strategies for attack that are ro-bust against the virus's capacity to escape *. Such knowledge can inspire targeted drug therapies and rational vaccine strategies that are aided by a deeper under-standing of virus sequence, structure and function [10, 11, 12, 13]. Considering HIV as an example, we demonstrate that such an understanding can be furnished by analyzing mutational patterns in patient-derived virus sequences and seeking evolutionary vulnerabilities that can be exploited to design vaccines that exploit these vulnerabilities. Chapters 2 and 3 address the following questions,
1. How might collective evolutionary constraints in viral proteins be inferred by examining mutational patterns in patient-derived viral sequences? (Chap-ter 2)
2. Can this information be harnessed to design immunogens (components of a vaccine) that induce cellular immune responses that limit potential escape routes during natural infection? (Chapter 2)
3. To what extent do mutational patterns in circulating viral strains, which are *"So in war, the way is to avoid what is strong and to strike at what is weak." (Sun Tzu, "The Art
sampled from diverse patients, accurately reflect intrinsic fitness constraints of the virus? (Chapter 3)
Despite the focus on HIV, the methodologies proposed to address these ques-tions can be applied to diverse RNA viruses for which sequence data is available. Identifying appropriate immune targets within viral proteins ("immunogen design") is but one of many aspects of a successful vaccination strategy [14]. Equally important is the understanding of the immunological mechanisms (in-volving both the innate and the adaptive arms) that mediate successful antivi-ral responses, and how these could be incorporated in the design of vaccines [15, 16, 17, 16, 18]. For example, analysis of HIV patient samples have shown that outside of their antigen specificity the proportion of cells executing multi-ple functions, rather than the bulk frequency of immune cells, correlates with effectiveness in slowing HIV disease progression [19, 20]. It is tempting to spec-ulate a vaccine designed to support the emergence of such functions can have a beneficial effect [16]. Studies in mice have supported this idea [21].
Recent advances in flow cytometry involving metal-labeled probes enable the quantitation of single-cell phenotypes involving > 40 parameters, including the constellation of cell-surface antigens, intracellular cytokines, transcription factors and regulatory enzymes, at the level of single cells [221. (Here, the phenotype of a cell is defined by its expression levels of each parameter within this constellation.) Phenotype dictates function. Thus, this provides an unprecedented opportunity to identify complex phenotypic subsets and their functional roles from patient samples; it is of particular interest to identify subsets whose frequency and pro-portion might be correlated with positive clinical outcomes. While associative,
the hypotheses emerging from such efforts can be tested in functional studies, and if validated, can directly instruct translational efforts.
Identifying complex phenotypes from high-dimensional cellular data is not a straightforward exercise, and conventional approaches relying on visual ex-amination of biaxial scatter plots are unscalable - as the number of measured parameters increases, number of such plots increase combinatorially. In Chapter 4, we introduce a computational tool rooted in nonlinear dirnensionality reduction that projects the high dimensional data onto a two-dimensional map, enabling automatic identification of phenotypic subsets and their visualization. In Chap-ter 5, we apply this tool to analyze expression data of immune cells sampled from HIV patients in search of features that correlate with control. Specifically we address the following questions,
1. How can T-cell phenotypes be discovered in an unbiased way from high dimensional single-cell protein expression data? (Chapter 4)
2. What phenotypic features of primary "helper" T cells (vide infra) derived from HIV infected patients correlate with natural viral control? (Chapter 5)
The rest of this chapter is organized as follows. Section 1.1 provides a short primer on adaptive immunity concepts. Sections 1.2-1.5 provide an extended context and background for each of the chapters. Section 1.6 is a short note on the concept of "dimensionality reduction", which is a recurring theme in this work.
1.1
The Adaptive Immune System
-
A primer
1.1.1
B cells and T cells
-
different arms of adaptive
immu-nity
Key players in adaptive immunity are two classes of cells - B cells and T cells. These cells arise from a common lymphoid progenitor cell through genetically encoded differentiation programs, and also go by the names B and T lympho-cytes respectively [3]. Each type of cell expresses a receptor on its surface that is central to its function - these are called the B and T cell receptor (BCR/TCR) re-spectively. Each B/T cell expresses many copies of the same BCR/TCR molecule, but these molecules are likely to be different across two B cells or two T cells. The specific receptor on each B/T cell is generated through somatic recombination of a set of germline-encoded genes (that are distinct for B cells vs. T cells) [23], and the combinatorial outcomes of this process give rise to an extraordinary clonal di-versity of B and T cells that circulate in the body. This didi-versity is the cornerstone of adaptive immunity [24, 25].
BCRs and TCRs bind to molecular signatures of pathogens (termed "epi-topes") that triggers the activation of these cells, and under appropriate con-ditions can initiate a systemic immune response. Occasionally, these cells can in-correctly activate against self-proteins, thereby initiating autoimmune responses that are detrimental to the host; this is the cause of diseases like multiple sclero-sis, type 1 diabetes and rheumatoid arthritis. To reduce the likelihood of autoim-mune responses, the germline-encoded diversity of BCR/TCR genes is consid-erably pruned through a series of mechanisms to ensure that receptors binding
strongly to self-proteins are deleted [26, 27]. These mechanisms are different for B and T cells [27].
On the other hand, BCRs (but not TCRs) can undergo additional somatic evo-lution during an immune response through a process called affinity maturation [28], where mutations are introduced in BCR genes over multiple rounds by the enzyme activation-induced cytidine deaminase (AID), and high affinity variants are selected successively. This remarkable process (akin to Darwinian evolution) occurs within areas of the lymph nodes called germinal centers [29, 30].
1.1.2
Different modes of recognition of pathogens and
im-munological memory
In addition to their distinct mechanisms of maturation, B cells and T cells also combat infection through different but complementary mechanisms. Upon ac-tivation, BCRs can be secreted into the blood as antibodies (Abs) that can bind to protein receptors on the surface of pathogens that circulate in the blood, and "neutralize" their capacity to invade new cells, thereby reducing the spread of in-fection. In contrast, T cells are involved in recognizing and eliminating host cells that are infected. T cells consist of multiple subsets that perform different func-tions during an adaptive immune response [31, 32, 33]. Prominent among these are "helper" and "killer" T cells, identified by their selective expression the co-receptors CD4 and CD8 on their surfaces respectively [34]. Multiple sub-lineages of these cells exist and carry different functional roles [32].
CD4+ T cells are key orchestrators of the adaptive immune response. They play an important role in activating other cells, including B cells, macrophages,
and other kinds of T cells, inducing them to acquire effector functions that collec-tively execute an immune response. CD8+ T cells recognize infected cells and in response, release cytotoxic molecules that kill the infected cell; activated CD8+ T cells that carry out cytotoxic function are called cytotoxic T lymphocytes (CTLs). T cells, unlike B cells, do not recognize whole proteins but peptide fragments derived from proteolytic degradation of these proteins. Specifically, TCRs bind to short peptide fragments (p) bound to protein products of the Major Histocom-patibility Complex (MHC) genes expressed on the surface of nearly all cell types in the body [35]. Specifically, CD8+ T cells recognize peptides bound to MHC class I molecules while CD4+ T cells recognize peptides bound to MHC class II molecules. Colloquially, CD8+ and CD4+ T cells are said to be "restricted" by MHC class I and class II respectively. While MHC class I proteins are expressed on every cell, MHC class II is expressed only by certain kinds of cells that serve as professional antigen presenting cells (APCs) - these include Dendritic cells, Macrophages, certain B cells and certain activated epithelial cells [3].
In the absence of infection, the pMHC complexes expressed on cellular sur-faces involve peptides derived from endogenous proteins; on the other hand, if a pathogen has invaded a cell, a few of these pMHC complexes will involve peptides derived from pathogenic proteins, which can be recognized by specific T cells leading through the initiation of a systemic immune response. Filtra-tion processes in the thymus ("thymic selecFiltra-tion") only allows those T cells to mature that discriminate efficiently between self- and pathogen-derived pMHCs [361. This recognition is also highly exquisitely sensitive - T cells can activate in response to a single pMHC in a "sea" of thousands of endogenous pMHCs [37]. Vaccines work by priming the immune response against attenuated forms of
the pathogen delivered to the host by inoculation. This induces memory B cells and T cells that circulate in the periphery, and are "waiting and ready to strike" during a real infection by the pathogen. The modern science of vaccination be-gins with the work of Edward Jenner t, the 1 8 th century English physician who
discovered that injecting individuals with an inoculum of the nonlethal cowpox virus made them immune to infection by smallpox, a major cause of death in that period. Advanced further by Pasteur, Hilleman, Salk and others, vaccination is one of the greatest triumphs of modern medicine that has saved many lives, leading to the near-eradication of once-debilitating diseases like smallpox and polio.
1.2
Collective evolutionary constraints in HIV
pro-teins and its relevance for rational
immuno-gen design
More than thirty years have elapsed since HIV was discovered to be the causative agent responsible for Acquired Immunodeficiency Syndrome (AIDS) [38, 39], but the prospects of a universal prophylactic vaccine continue to remain un-certain [40]. More than 35 million people across the world live with HIV/AIDS at present and the pandemic claimed 1.6 million lives in 2012 [41]. The burden of HIV/AIDS is greatest in sub-Saharan Africa, where in a large proportion of
tThe concept of variolation, i.e. inoculation with pustular material from an infected survivor to elicit immunity in an uninfected person appears to have many historical antecedents, long before Jenner's work. See M. Lombard, P. P. Pastoret and A. M. Moulin, Rev. Sci. Tech, 26(1):29-48, (2007) for a fascinating review
areas more than two-thirds of the resident population is infected.
1.2.1
In vivo characteristics of HIV infection
HIV primarily infects CD4+ helper T cells, macrophages and dendritic cells [42, 43]. Following an acute phase of infection with flu-like symptoms, patients transition into a largely asymptomatic chronic phase, characterized by a steady viral load measurable in the patient's blood and lymphatic tissue (the so called "set-point viral load" or SPVL). During the acute phase more than half of the res-ident CD4+ T cells get depleted in the gut and other lymphoid tissues, an insult that foreshadows the slow but steady debilitation of the immune system during the chronic phase, ultimately leading to systemic immunodeficiency [44]. The CD4+ T-cell count is the most important measure used in disease prognosis. The length of the asymptomatic phase and SPVL exhibit a high degree of variation among HIV infected patients. However, the SPVL is strongly anti-correlated with the length of the asymptomatic phase, and is also used as a prognostic indicator [45].
Quiescent on the surface, the asymptomatic phase is in fact highly dynamic, wherein half the viral population turns over a timescale of two days [46]; such a high replicative fecundity is the principal source of mutations that evade immune responses in vivo. Ab or TCR mediated-recognition of viral epitopes is highly specific and therefore can be abrogated by a single point mutation within the epitope [47, 48]. These mutations lead to "viral escape", and a new wave of T and B-cell responses are initiated against the escape variant [49, 50]; it is often only a matter of time until escape variants against these new responses arise and
render them ineffective. In the absence of therapeutic interventions this battle, set against the background of slow depletion of CD4+ T cells, ultimately leads in severe immunodeficiency in the host. This state is characterized by extremely low CD4+ T cell counts (< 200 cells/pl). Immune function is compromised to such an extent that the host is finally killed by opportunistic infections.
Like all retroviruses, HIV exhibits error-prone replication [51]. When HIV in-fects a target cell, its RNA genome is reverse-transcribed into the complementary DNA molecule by the viral enzyme reverse-transcriptase (RT), a RNA-dependent DNA polymerase. RT has a replication error rate of approximately 2-4 x 10-5 per base per replication cycle; although the replication machinery of many retro-viruses exhibits a similar error rate, the high sequence variation seen in HIV patients is primarily facilitated by its rapid turnover in vivo [46].
Within a host, the viral population exists as a cloud of closely related mu-tants in sequence space (the so-called "quasispecies") [52, 53], and early work indicated that this variation is not reflected in individual isolates of viral samples [54]. Compared to the "consensus" (average) strain, the quasispecies contains all the viable strains separated by a single mutation, and a large proportion of viable strains separated by two mutations. Sequencing studies have found that the quasispecies diversity of HIV within a single infected individual sampled at a single time point is comparable to the global diversity of influenza strains sampled during a pandemic year [9].
Implications of HIV mutability for therapies and
vac-cines
The existence of such formidable sequence diversity in vivo implies that antiretro-viral monotherapy is likely to be thwarted by the selection of resistant strains that are likely to exist within the quasispecies reservoir [46, 55, 56]. This early real-ization led to the recommendation of multi-drug cocktails (typically three drugs) to treat HIV patients; the invention of antiretroviral therapy (ART) has been sin-gularly responsible for making the epidemic manageable in developed countries [57], and has also been responsible to avert hundreds of thousands of deaths in developing countries through emergency relief programs like the United States' PEPfAR program [58]. According to the 2013 UNAIDS report, the scale-up of ART averted over 5.4 million deaths between 1994 and 2012.
While these drugs afford many patients the ability to lead normal lives with undetectable viral levels in the blood, they do not kill infected CD4+ T cells that are inactive. In these "latently-infected cells", the proviral DNA is not actively transcribed [59]; this latent reservoir of HIV decays with a mean half-life of ~ 4 years [60]. When taken off therapy after many years, the viral loads in patients have been found to jump to high levels in a matter of weeks due to the
re-activation of latent cells.
Despite these laudable efforts, public health statistics suggest that for every two patients placed on combination ART, five people become infected. Therefore, only a universal vaccine can be a long term solution to this global health crisis [61].
Traditional vaccines employed whole proteins of the pathogen as
gens (vaccine components), and this strategy was successful for pathogens that exhibited significantly lower mutational diversity compared to viruses like HIV and HCV. Using whole proteins primes the natural immune response, and this strategy sufficed when the pathogen in question did not exhibit high sequence diversity. Despite its terrorizing morbidity, the rate of fatality associated with smallpox was only 30 % [3]; 70 % of infected individuals successfully cleared the virus, suggesting that an early induction of immune responses through vac-cination should sufficiently empower the immune system. In contrast, in the 32 year history of the HIV pandemic, there has not been a single reported case of a patient that has successfully cleared the infection through natural immune mechanisms.
When this fact, and the extreme mutability of HIV described earlier are taken into account, it should not come as a surprise that vaccination trials based on traditional immunogenic formulations that prime natural immune mechanisms have not yielded success [62]. In the STEP trial, the Merck Adenovirus serotype (Ad)5 viral-vector expressing HIV proteins Gag, Pol, and Nef failed to improve control in vaccinated subjects; worse, there was increased acquisition of HIV in the vaccinated group, although the reason for this result was ascribed to preex-istence of Ad5 vector-specific Abs in some participants and lack of circumcision in males [63].
It is now well accepted that an efficacious HIV vaccine must be able to induce immune responses with special properties - Abs with a high degree of cross-reactivity towards mutational variants ("broadly neutralizing" Abs or bnAbs), and also CD8+ T-cell responses that recognize parts of the virus where mutation-induced escape is less likely to emerge [64]. bnAbs exist in low concentrations
in ~ 20% patients, but are highly somatically mutated compared to conventional Abs [65]. A recent therapeutic study involving a macaque model of HIV infection found that administration of small doses of these antibodies can bring about rapid albeit temporary reduction in viremia to undetectable levels, and future studies might evaluate their utility in a therapeutic vaccine setting [66]. The prospects of eliciting bnAbs through vaccination, however, remain unclear.
1.2.3
Importance of CD8+ T-cell responses against HIV, and
implications for a T-cell vaccine
A great deal of understanding about the role of T cells in influencing in vivo dynamics of HIV has emerged from studies of infected patients and experimental models of HIV infection involving non-human primates. This section reviews some of these findings.
Studies in humans
Clinical studies in HIV patients have revealed that MHC class I-restricted CD8+ T-cell responses play a critical role in driving viral evolution longitudinally within a patient [49], and at the population level [67]. One of the earliest findings that is still not fully understood, is that robust CTL responses with effector function are detectable in HIV-infected patients despite CD4+ T-cell responses being severely impaired [68]. Sequencing of autologous virus from infected persons revealed adaptive mutations supporting the positive selection pressure mediated by CTL induced-immune responses [69]; recent studies focusing on the acute phase have confirmed that CTL induced escape occurs within 2-4 weeks of infection [49].
The strongest evidence that CTLs can contribute to positive clinical outcomes has come from the discovery of "elite controllers", patients that maintain ex-tremely low set point viral loads (SPVLs) undetectable even by ultrasensitive assays (< 50 copies/ml) in the absence of any therapy [70]. These individuals are highly enriched for certain MHC class I alleles, in particular HLA-B*5701, HLA-B*5703, HLA-B*2705 and in african ethnicities, HLA B*5801, implicating a role for CD8+ T cells in viremic control in these patients f. Furthermore, plasma virus sequenced from elite controllers have been found to contain CTL escape mutations that are associated with decreased replicative capacity, suggesting that the natural immune responses in these patients target parts of the virus, where escape comes at a cost to fitness [71, 72]. It has also been noted that the charac-teristics of thymic development in Elite Controllers result in a higher proportion of cross-reactive CD8+ T-cells in the periphery [73]. Such T-cells possess TCRs that are more resistant to point mutations in viral eptiopes.
These genetic associations with SPVL have been confirmed by population wide clinical studies across multiple cohorts of patients [74, 75]. In particular, in a recent genomewide association study (GWAS) involving 300 groups had analyzed data from 974 HIV controllers, who maintained SPVLs less than 2000 copies/ml in the absence of therapy, and 2648 HIV progressors with SPVLs in ex-cess of 50,000 copies [75]. Analysis of correlations between viral load and more
tThe MHC locus in the human genome is referred to as the Human Leukocyte Antigen (HLA). The HLA class I locus on each of the two human karyotypes (a single set of chromo-somes) is specified by a "haplotype" of three genes termed HLA-A, HLA-B, HLA-C. Within each karyotype, these three genes are tightly linked by linkage disequilibrium and are present on chro-mosome 6. An individual thus possesses six HLA class I genes, two each of A, B and C alleles. The HLA class II locus haplotype is also present on chromosome 6, but possesses a different nomenclature. The HLA class I and class II loci are the most polymorphic in the entire human genome, based on the diversity of alleles that can be present across individuals in a population.
than a million single nucleotide polymorphisms (SNPs) revealed over 300 sta-tistical associations, and remarkably all of these lay within the HLA region of chromosome 6. Only four SNPs survived correction, however, for multiple hy-pothesis testing (which reduces the incidence of false positive results), and two of these were markers for HLA-B57 and HLA-C expression respectively, consis-tent with the previous GWAS study [74]. Thus evidence from rare instances of natural infection suggests that CTLs can control viremia, but these mechanisms do not lead to clearance of the virus.
An important caveat is that these genetic associations explain only -13% of the variance in the SPVL in the data (this increases up to 22 % if age and sex are included) [74] -i.e. a large fraction of individuals possessing these so-called pro-tective HLA alleles do not end up as controllers. An important future direction of HIV research is to integrate large scale host genomic, transcriptomic and viral sequence datasets to gain better understanding of what factors (viral and host) contribute to these differences in clinical outcome continues [76].
Studies in non-human primates
The role of CTLs in HIV control is also supported in models of non-human pri-mates. When indian rhesus macaques are infected with Simian Immunodefi-ciency Virus (SIV, a virus that shares a common ancestor with HIV) or SIV-HIV hybrid virus (SHIV), they develop acute viremia progressing to chronic infec-tion and finally develop AIDS-like symptoms (concomitant with a massive loss of CD4+ T helper cells), similar to humans infected with HIV [77]. Similar to humans, certain macaque MHC alleles are strongly associated with viral control (e.g. Mamu B*08), and intriguingly many of these alleles have been found to be
homologous to human alleles that are enriched in controllers (e.g. Mamu B*08 is homologous to HLA B*27) [78]. Selective ablation of CTLs in macaques in-fected with SIV through CD8-specific monoclonal Abs leads to a rapid increase in viremic levels, providing direct support of an antiviral role for CTLs [79].
Despite important physiological differences from HIV infection in humans, the macaque model has been an intense testing ground for vaccination and thera-peutic concepts [80, 81, 82, 83, 84, 85, 66]. Early results indicated that vaccines that stimulate CTLs lead to enhanced viremic control in rhesus macaques challenged with SIV or SHIV. Recent results employing a novel vaccination approach, which involves an unconventional rhesus monkey cytomegalovirus (RhCMV) vector ex-pressing recombinant SIV genes as the immunogen, have reported the elicitation of potent effector memory responses in the vaccinated monkeys [81, 84, 85]. Af-ter being challenged with pathogenic SIV strain, all vaccinated subjects were infected, but 50% cleared the virus soon after the acute phase. In the mon-keys that cleared virus, selective ablation of CTLs did not cause a rebound in viremia [84, 85]. Surprisingly, this CMV based vaccine elicited highly atypical T-cell responses that had thrice the epitope breadth of conventional vectors; also two-thirds of the CD8+ T-cells were restricted by MHC class II. Despite these caveats, this unprecedented study has renewed vigor in the hope for an T-cell vaccine for HIV. Taken together, the results from the macaque model confirms the important role of CTLs in controlling HIV infection, lending support to the idea of a putative T-cell based vaccine against HIV.
The immunogen design question
Compared to laboratory settings in SIV vaccination studies that typically use specific strains of the virus, vaccine design against HIV has to contend with the immense diversity of circulating sequences that can initiate infection [86]. In addition, HIV vaccines have to confront the high mutability of the virus in vivo, and induce responses that limit possibilities of escape for the virus. Kiepiela et al. found from their analysis of T-cell responses in a large clinical cohort that while a higher number of Gag-specific T-cell responses were associated with control (Gag - a highly conserved polyprotein, which encodes the structural components of HIV), a higher number of Env-specific T-cell responses were associated with progression (Env - the protein which forms the HIV receptor, known to exhibit high sequence variability) [87].
Vaccines that prime responses against conserved proteomic regions have been proposed as solutions [11, 88, 89] .In this framework, linear portions of HIV pro-teins that exhibit a high degree of average single-site conservation are selected as immunogens. The underlying rationale is that such regions are important for viral viability, and therefore most amino acid mutations in such regions (that the virus would need to make in order to escape an immune response) are likely to compromise viral fitness [11, 90]. Priming a T-cell response towards such re-gions is therefore, the argument goes, likely to be resistant to possible mutational escape routes.
This approach suffers from an important limitation in that mutational escape routes are inherently nonlinear and involve "epistatic" interactions between mul-tiple residues [91, 92, 93, 94]. Indeed, it has been clinically observed during the
natural immune response that while a primary mutation that escapes CTL pres-sure confers a fitness cost on viral replication, compensatory mutations emerge from distal parts of the genome that restore viral replicative fitness [95]. It follows from these observations that an efficacious vaccine must minimize the possibility of such collective routes to escape and this can be accomplished by targeting pro-teomic regions wherein a greater proportion of strains carrying multiple muta-tions are unviable. However, measures such as "average single-site conservation" do not account for such collective effects that involves multiple mutations acting in concert.
Thus, we hypothesized that characterizing such epistatic interactions might help identifying such regions in HIV proteins, and thus lead to a further refine-ment of the "conserved vaccine" concept, where conservation is defined based on multidimensional constraints rather than uni-dimensional constraints. Chapter 2 describes the analysis of collective mutational correlations within HIV pro-teins using Random Matrix Theory (RMT), which was originally developed in high energy physics but has since then been applied to diverse realms such as finance and biology. Based on our results we propose immunogens containing multidimensionally conserved proteomic regions that are optimized to maximize responses in the Caucasian American ethnic group.
1.3
Faithful inference of intrinsic fitness landscapes
from patient-derived viral sequence data
1.3.1
What are viral fitness landscapes?
The computational approach delineated in Chapter 2 identifies proteomic regions that exhibit statistical signatures of collective mutational vulnerability. This qual-itative approach, however, lacks the ability to quantify precisely the fitness effect of specific combinations of mutations, or even determine whether one particular combination of mutations has a higher positive or negative impact on viral fitness compared to another. Answers to such questions can be furnished by knowing
the fitness landscapes of proteins of the virus [96, 97, 98, 99].
Thefitness landscape was first postulated as a conceptual tool by Sewall Wright
in seminal work concerning evolutionary genetics [100]. In our context, knowing the fitness landscape of a viral protein is equivalent to assigning a measure of replicative fitness (vide infra) to each unique mutant strain of the protein [96]. Such a comprehensive knowledge would reveal specific regions of the proteins vulnerable to immune targeting, providing useful information for the design of immunogens that induce efficacious T/B-cell responses [98]; furthermore, knowl-edge of the fitness landscape would also enable the computational prediction of the likely escape paths, and incorporate additional responses that block these.
Varied interpretations of the word "fitness" abound in literature - it could be a property of an individual organism or of a collective, and changing the
envi-Implicit is the proviso that proteins evolve independently or are only weakly coupled to each other. If this assumption is not valid, then whole-proteome fitness landscapes must be sought.
ronment often changes fitness considerations. It is often defined as a propensity or probability to produce a certain number of offspring [101]. In this work, the word "fitness" is largely used in a narrow context to imply "replicative capacity (RC)" of a particular mutant strain, and to emphasize this point we often refer to it as intrinsic fitness. RC is defined as the mean number of cells infected by an infected cell with unbounded access to target cells, and in the absence of ex-ternal factors like drugs and immune responses. RCs of specific strains can be measured in standard laboratory assays (e.g. GFP based assay described in [94]).
1.3.2
Inferring fitness landscapes from data
One approach to infer fitness landscapes from data is through the fitting of mathematical models to diverse in vitro measurements of RC for diverse strains through regression [96, 97]. Such an approach, however, requires a large number of expensive and laborious experiments. An alternative approach proposed by Ferguson et al. [98] and expanded in Mann et al. [102] is the use of compu-tational inference methods rooted in entropy maximization to construct fitness landscapes based on datasets [103] of circulating viral sequences that are sam-pled from diverse patients . This approach rests on a simple ansatz : strains that
are intrinsically fitter must also be more prevalent in the population when averaged over a large diversity of immune backgrounds from which these strains are sampled. Thus
the prevalence landscape, the argument goes, must somehow reflect the intrinsic fitness landscape.
But what is the computational procedure underlying this inference? Briefly, the sequence data for a particular protein contains information on the probability
of occurrence of specific amino acids at each site, the probability of occurrence of specific doublets at each pair of sites, and similarly, the probability of occurrence of a specific combination of n amino acids at n specified sites for n > 3. It can be shown that a prevalence model that maximizes the likelihood of observing the sequences in the data also reproduces these correlations [98]. Assuming that the data is well-sampled, this prevalence model describes the statistical properties of the evolutionary space spanned by real protein sequences.
However, since the number of available sequences is much smaller than the possible combinations of amino acid doublets, triplets, quadruplets etc. even for a modestly sized protein (- 100 residues), it is impossible to fit a model that reproduces the correlations at all orders. One can invoke the maximum entropy principle to seek the least biased model that reproduces only some of the low order correlations, say the probabilities of amino acid occurrences at the single site and the two-site level [104, 105]. The resulting model can then be used to predict higher order moments and other quantities reflected by the data that were not included in the fitting procedure. Such an approach has been previously applied to infer networks from neuronal activity data [106] and to model antibody diversity [107].
The maximum entropy inference leads naturally to a model of prevalence where the probability of a particular amino acid sequence is described by a Boltz-mann distribution governed by a Hamiltonian ("energy") whose value depends on the sequence [98]. The form of the Hamiltonian is reminiscent of spin mod-els in statistical physics, and its parameters (including fields and coupling con-stants) are fitted such that the model reproduces the low order correlations in the sequence data through numerical procedures. The problem of inferring the
fields and coupling constants from the correlations is referred in the literature as the "Inverse Inference problem", the computational issues involved are suf-ficiently non-trivial to have sparked a flurry of activity [108, 109]. In Ferguson et al., viral protein sequences were converted to a binary representation similar to that introduced in Chapter 2, such that at each site '0' represented the "wild type" (most frequent) amino acid while '1' represented any mutant [98]. Because of this binary approximation, the Hamiltonian of the resulting prevalence model represents an Ising system. In contrast, Mann et al. extended this framework to explicitly consider the amino acid identities of mutations, which leads to Hamil-tonians resembling Potts models [102]. In either case, the model predicts that the energy of a strain inversely correlates with its prevalence.
1.3.3
Agreement with RC measurements and the puzzle
The quality of the inferred fitness landscape was tested on its ability to repro-duce other statistical quantities measurable in the sequence data that were not used as part of the fitting procedure, and finally experimental measurements of
in vitro replication capacity. Ferguson et al. constructed prevalence landscapes
for structural proteins of HIV and showed that these were accurately able to predict fitness relations among diverse HIV strains in in vitro replication assays [98]. Their predictions also tested positively against in vivo sequence evolution of HIV strains when the immune responses were known. This suggested that the inferred "prevalence landscape" is a suitable proxy for the "intrinsic fitness landscape".
Each sequence sampled from the set of circulating sequence in the population was the result of adaptation to a pattern of immune response determined by a particular host's HLA genetic background; these responses are highly diverse across different individuals in a population [110]. Indeed, HLA-B is the most polymorphic locus in the entire human genome [111]. Furthermore, almost all of this variation is concentrated among residues that line the peptide binding groove in these molecules [112] -thus the existence of HLA molecules that bind diverse peptides is of sufficient functional importance to be subjected to Dar-winian selection. How do diverse within-host immune responses shape the evo-lution of HIV at the population level? Do the statistics of mutations reflected in prevalence data correctly reflect properties of the intrinsic fitness landscape?
Chapter 3 explores this question by simulating viral population dynamics in individual patients that form a sexual network - our scheme integrates within-host quasispecies dynamics on fitness landscapes with within-host-to-within-host transmission. The simulation results are interpreted by the analysis of a statistical mechanical model that mimics essential features of our simulations, and we derive an ap-proximate analytical relationship that elucidates the connection between preva-lence and fitness.
1.4
Identification of cellular phenotypes from high
dimensional expression data
1.4.1
Measuring single-cell phenotypes
The invention of fluorescence activated cell sorting (FACS) technology in the 1970s enabled for the first time phenotypic analysis of individual cells in cellular populations, which transformed the field of immunology [113, 1141. In FACS, monoclonal Abs that specifically bind proteins of interest are conjugated with fluorophore molecules that emit light signals in a narrow range of wavelength when excited by a laser probe. Cells are individually probed by means of a flow-based sorter, and the intensity of the emitted light is a quantitative measure of the amount of protein. Conjugating each type of antibody with a fluorophore with a distinct emission wavelength allows the simultaneous measurement of multiple proteins. FACS technology enabled immunologists to discover different immune cell phenotypes and the developmental relationships between them [114, 115, 116, 117]. Multiparameter flow cytometry is now being applied to characterize the quality of immune response following immunizations with various vaccines and also after viral infections [118].
FACS is, however, typically limited to measuring 12 parameters simultane-ously. Increasing the number of probes can result in an overlap between their emission spectra, which introduces difficulties in resolving the quantity of each protein being measured requiring "compensation" for spectral overlap [119, 118]. This limitation has been overcome by mass cytometry, which currently allows for
the simultaneous measurement of > 40 proteins, a number that can in theory be increased up to 100 [22]. In mass cytometry, antibodies are conjugated with rare earth metals (one for each type of probe). During measurement, each individual cell is nebulized and the quantities of different rare earth metals, each of which reflects the quantity of one of many proteins of interest, are measured on an inductively coupled mass spectrometer. While still a relatively new technology, mass cytometry has been shown to be comparable to FACS on most performance metrics [22, 120].
1.4.2
Computational challenges involving the analysis of
high dimensional data
The ability to measure dozens of parameters at single cell resolution has opened up exciting new possibilities for phenotypic analysis of cell populations at an unprecedented depth. Since phenotype governs function, it is hoped that these efforts would advance our understanding of functional relationships of different cell types in general, and within the immune system in particular. As the technol-ogy is still in its nascency, a number of studies have applied it to well-known bi-ological scenarios, as it makes it easy to interpret and validate the results. For ex-ample, Bendall et al. analyzed human bone marrow cells for the expression of 30 proteins (chosen on the basis of prior biological knowledge), and showed using a clustering technique that the resulting high dimensional data could be used to re-construct lineage relationships between cell types in hematopoesis [120]. Newell et al. used mass cytometry to analyze primary human CD8+ T cells, and using principal component analysis (PCA) showed that well known CTL phenotypes