HAL Id: tel-01587772
https://tel.archives-ouvertes.fr/tel-01587772
Submitted on 14 Sep 2017HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
cancers, sarcomes et mélanomes pédiatriques.
Fanélie Jouenne
To cite this version:
Fanélie Jouenne. Apport du séquençage d’exomes constitutionnels dans l’identification de nouveaux gènes de prédisposition aux cancers, sarcomes et mélanomes pédiatriques.. Cancer. Université Paris Saclay (COmUE), 2017. Français. �NNT : 2017SACLS224�. �tel-01587772�
NNT : 2017SACLS224 Thèse de doctorat de L’Université Paris-Saclay préparée à Gustave ROUSSY Ecole DOCTORALE N° 582
Cancérologie : biologie – médecine - santé
Spécialité de doctorat : Sciences de la vie et de la santé
Par
Fanélie JOUENNE
Apport
du séquençage d’exomes constitutionnels dans l’identification de
nouveaux gènes de prédisposition aux cancers, sarcomes et mélanomes
pédiatriques
Thèse présentée et soutenue à Gustave Roussy, le 31 août 2017 à 16h
Composition du Jury :
Monsieur le Professeur Jean-Yves SCOAZEC Président Monsieur le Professeur Pierre VABRES Rapporteur Monsieur le Professeur Nadem SOUFIR Rapporteur Monsieur le Docteur Jacques GRILL Examinateur Monsieur le Docteur Marc-Henri STERN Examinateur Madame le Docteur Brigitte BRESSAC-de PAILLERETS Directeur de thèse
REMERCIEMENTS
Tout d’abord, j’adresse mes remerciements aux membres du jury qui ont accepté de juger cette thèse, les Professeurs Nadem Soufir et Pierre Vabres en tant que rapporteurs, qui ont apporté leur jugement éclairé à ce travail, les Docteurs Jacques Grill et Marc-Henri Stern comme examinateurs et le Professeur Jean-Yves Scoazec qui a accepté de présider ce jury.
Je tiens à remercier ma directrice de thèse, le Docteur Brigitte Bressac-de Paillerets, pour m’avoir accueillie dans son équipe à Gustave Roussy. Ces six années passées à vos côtés m’ont beaucoup apportées, je regrette que l’histoire s’arrête là et j’espère avoir l’occasion d’échanger à nouveau avec vous, comme lors de nos nombreuses réunions de travail. Vos connaissances et votre passion m’ont toujours émerveillées et ont été une vraie stimulation.
Je remercie également toute l’équipe du service de génétique et celle de pathologie moléculaire. Merci d’avoir toujours répondu à mes questions et su me divertir quand j’en avais besoin! Un merci particulier à Odile, pour son écoute et ses conseils tant d’un point de vue professionnel que personnel! Un grand merci à Vincent pour m’avoir accompagné tout au long de ce parcours. Et merci Mahaut, qui m’a tant appris et a quitté l’équipe trop vite.
Je remercie l’équipe de l’UMR118 et le Docteur Fathia Mami-Chouaib, pour leur accueil dans leur équipe pour mes répétitions de thèse. Je remercie également Jean Feunteun pour sa disponibilité et ses conseils.
Merci à ma nouvelle équipe de Saint Louis, pour avoir supporté mes « humeurs » et mes moments d’absence (physiques ou intellectuels !!) sur la dernière ligne droite de cette thèse ; spécial mention à Baptiste, en 3 mots : PDF-word-merci ;-). Je remercie le Professeur Samia Mourah pour m’avoir laissé du temps pour terminer ce travail et pour son accueil pour cette nouvelle aventure dans le somatique !
Mes remerciements vont ensuite à ma famille et à mes amis. A ma mère, Françoise, « recommence »…
A mes frères, Antoine et Jean-Baptiste et ma belle-sœur, Julie, je ne suis pas sûre que vous compreniez toujours ce que je fais, mais de vous avoir à mes côtés, dans ma tête et dans mon cœur, constitue une force qui m’a permis de faire tout ça, alors merci !
A mes neveux, Gaston et Marcel, leurs sourires, leur innocence et leur amour sans condition ont permis de me conduire jusqu’ici.
A ma nouvelle famille, la famiglia, Sandro, Flavio, Charlène, Giovanni, Matteo, Giulia et Abella, merci pour tous ces bons moments de détente, à la Rochelle ou en Gwada, nécessaires pour recharger les batteries.
A la famille que je vois peu mais qui compte tant : ma Moumoune, mon parrain Pierre, mon oncle Jean-Jacques...
A Audrey, mon amie, ma 1/2B, quel chemin parcouru! Depuis nos années fac, où on révisait nos cours entre 2 apéros, qui aurait dit que tant d’années plus tard, j’en serais là. Et pendant tout ce
temps, tu étais là! Et aujourd’hui, je ne peux imaginer que j’en serais arrivé là sans toi, ton soutien, ton amitié et tous nos moments.
A Jean-François, à Christine, si loin mais toujours là !
Aux bordelais toujours présents malgré la distance : ma bouli Karine, ma Geri, Leslie, Juju et ses juju, Pauline, Benoit, …
A toutes mes rencontres parisiennes : Lauriane, Sylvie, Sylvine, Laeti, Coco, Fabien, Sabrina, Valentine … et mes coups de cœur parisiens : Ophélie, Michael, ma rouquinette Capu, Guillaume-Francis. Aux potes « imposés » mais tant appréciés : Marie-Héléne, Vincent, Ariane, Wil, Bruno, Bénédicte, Christian, … . Et à tous ceux que j’oublie…
Et enfin à toi, Gianni…on a réussi!! Merci infiniment pour ton soutien, je n’aurais jamais vu le bout de cette thèse sans toi! Combien de soirées, de week-ends, de journées où tu as tout géré comme un chef pour me permettre de me consacrer à ce travail ! Merci merci merci !!! Et maintenant que tu m’as permis d’en arriver là, faisons en sorte que le Docteur Jouenne devienne, Docteur Vitillo !
LISTE DES ABREVIATIONS
-MSH : Melanocyte-stimulating hormone ACD : adrenocortical dysplasia protein homolog ACOT13 : acyl-CoA thioesterase 13
ADN : Acide désoxyribonucléique AKT : AKT Serine/Threonine Kinase 1
ALDH1L1 : aldehyde dehydrogenase 1 family member L1 ARF : Alternative Reading Frame
ARFIP1 : ADP ribosylation factor interacting protein 1 ARHGEF5 : Rho guanine nucleotide exchange factor 5 ARID1A : AT-Rich Interaction Domain 1A
ARID2 : AT-Rich Interaction Domain 2
BAP1 : Breast cancer 1 (BRCA1) associated protein 1 BRAF : B-Raf Proto-Oncogene, Serine/Threonine Kinase CADM2 : Cell Adhesion Molecule 2
CDK4 : Cyclin-dependent kinase 4
CDKN2A : Cyclin-dependent kinase inhibitor 2A CDKN2B : Cyclin-dependent kinase inhibitor 2A CGH : Comparative Genomic Hybridization CNG : Centre National de Génotypage
COSMIC : Catalogue Of Somatic Mutations In Cancer
CRISPR/Cas9 : Clustered regularly interspaced short palindromic repeats/CRISPR associated protein 9 CTLA-4 : Cytotoxic T lymphocyte antigen 4
CRB : centre de ressources biologiques CSD : chronically sun-damaged
dbSNP : Single Nucleotide Polymorphism database DCT: Dopachrome Tautomerase
DSCAM : DS cell adhesion molecule
EIF4G2 : Eukaryotic Translation Initiation Factor 4 Gamma 2 EMT : Transition épithélio-mesenchymateuse
ESP : Exome Sequencing Project EVS : Exome Variant Server
ExAC : Exome Aggregation Consortium FBXO32 : F-box protein 32
FFPE : formalin-fixed paraffin-embedded FISH : hybridation in situ en fluorescence
GIST : tumeur stromale gastro-intestinale - gastrointestinal stromal tumors GNA11 : G protein subunit alpha 11
GNAQ : G protein subunit alpha q GR : Gustave Roussy
GRM4 : Glutamate Metabotropic Receptor 4 GST : Gènes suppresseurs de tumeurs GWAS : Genome Wide Association Study HHV8 : Human Herpes Virus
HRAS : HRas proto-oncogene, GTPase IHC : immunohistochimie
INK4a : Cyclin-Dependent Kinase Inhibitor 2a ISKS : International Sarcome Kindred Study IVA : Ingenuity Variant Analysis
LOF : Loss of fonction LOH : Loss of heterozigosity
MAP Kinase: Mitogen-activated protein kinase MATP: membrane associated transporter protein Mb: mégabase
MC : mélanome cutané
MC1R: Melanocyte-stimulating hormone receptor 1 MDM2: Mouse Double Minute 2
MEC : matrice extracellulaire
MEK: Mitogen-activated protein kinase kinase - MAPK/ERK kinase MITF: Microphtalmia-associated transcription factor
MLH1: MutL Homolog 1
MMP : matrix metalloproteinase
MPNST : tumeur maligne des gaines des nerfs périphériques MSH : melanocyte stimulating hormone
MSH2: MutS Homolog 2 MSH6: MutS Homolog 6
MYBBP1A : Myb-binding protein NF1: neurofibromatose 1
NGS : next-generation sequencing NRAS : NRAS Proto-Oncogene, GTPase PD-1: Programmeddeath-1
PDGFRA : Platelet Derived Growth Factor Receptor Alpha PI3K: phosphatidylinositol 3-kinase
PIGU : phosphatidylinositol glycan anchor biosynthesis class U PMS2: PMS1 Homolog 2
POLE: DNA polymerase epsilon POMC : proopiomelanocortin POT1: Protection of telomeres 1 PRSS3 : protease, serine 3
PTEN: Phosphatase and tensin homolog
RASSF1A: Ras Association Domain Family Member 1 RB1: Retinoblastoma 1
RCP : réunion de concertation pluridisciplinaire RECQL4 : RecQ Like Helicase 4
ROBO1 : Roundabout Guidance Receptor 1 ROBO2 : Roundabout Guidance Receptor 2 ROS : Reactive oxygen species
RTK : récepteur tyrosine kinase
SCF : Skp, Cullin, F-box containing complex
SDHA : succinate dehydrogenase complex flavoprotein subunit A SF3B1 : splicing factor 3b subunit 1
SKA3 : Spindle And Kinetochore Associated Complex Subunit 3
SMARCA2 : SWI/SNF Related, Matrix Associated, Actin Dependent Regulator Of Chromatin, Subfamily A, Member 2
SNP: Single nucleotide polymorphism ou polymorphismes de nucléotide simple SOX10: SRY-Box 10
SSM : Superficial spreading melanoma.
STAT : Signal Transducer And Activator Of Transcription SWI/SNF : SWItch/Sucrose Non-Fermentable
TERF2IP: TERF2 interacting protein TERT: Telomerase Reverse Transcriptase TP53: Tumor protein P53
TSC1 : TSC complex subunit 1 TSC2 : TSC complex subunit 2 UV: ultraviolets
VCF : Virtual Card File
WES: Whole Exome Sequencing WGS: Whole Genome Sequencing WHO : World Health Organization XP : Xeroderma Pigmentosum
Table des matières
LISTE DES ILLUSTRATIONS ... 1
LISTE DES TABLEAUX ... 2
I.
INTRODUCTION ... 5
1.
La cancérogénèse moléculaire ... 5
2.
La prédisposition génétique ... 11
II.
CONTEXTE ET OBJECTIFS DES RECHERCHES ... 17
III.
PARTIE I : CAS DE FAMILLES À CAS MULTIPLES - FAMILLE À CAS MULTIPLES DE
SARCOMES ... 19
1.
Le sarcome ... 19
A. Définition ... 19
B. Formes cliniques et histologiques ... 19
C. Incidence et mortalité ... 21 D. Facteurs de risque ... 23 1. Facteurs environnementaux ... 23 2. Facteurs héréditaires ... 25 E. Diagnostic ... 27 a) Clinique ... 27 b) Morphologie et immunohistochimie ... 29 c) Biologie moléculaire ... 29 F. Traitement ... 31
2.
Résumé de l’article ... 33
3.
Article en ligne ... 39
4.
Travaux complémentaires non publiés ... 47
a) Etude fonctionnelle des variants PDGFRA ... 47
b) Hybridation génomique comparative (CGH) de sept cas de sarcomes CDKN2A+ ... 49
c) Nouveaux gènes candidats modificateurs du risque de sarcomes ... 49
5.
Discussion ... 53
6.
Conclusion et perspectives ... 59
IV.
PARTIE II : CAS DE CANCERS CHEZ L’ENFANT JEUNE – LES TRIOS MÉLANOME DE
L’ENFANT/PARENTS SAINS ... 61
1.
Le mélanome ... 61
A. Définition ... 61
B. Les mélanocytes ... 61
1. Les différentes couches de la peau ... 61
3. Biologie du mélanocyte ... 65
4. La transformation mélanocytaire ... 67
C. Epidémiologie ... 71
D. Facteurs de risque du mélanome ... 73
1. Facteurs environnementaux ... 73
2. Facteurs de susceptibilité individuelle ... 75
3. Facteurs génétiques et épigénétiques ... 77
E. Principaux types histologiques de mélanomes ... 91
F. Dépistage et Diagnostic ... 95
G. Classification pTNM ... 97
H. Prise en charge thérapeutique ... 97
2.
Contexte des recherches ... 103
3.
Hypothèses ... 107
4.
Objectifs ... 107
5.
Population étudiée ... 107
6.
Résultats ... 109
7.
Discussion et perspectives ... 121
V.
DISCUSSION - CONCLUSION GENERALES ... 125
VI.
REFERENCES ... 129
1. Références ... 129
2. Références bibliographiques ... 129
VII.
ANNEXES ... 141
1- SUPPLEMENTS DE L’ARTICLE EN LIGNE ... 141
2- ARTICLES EN CO-AUTEURS ... 161
3- MÉTHODES ... 161
1. Extraction d’ADN ... 161
2. Préparation des échantillons tumoraux ... 161
3. Bases de données et logiciels utilisés ... 163
4. Séquençage à haut débit, séquençage d’exomes ... 167
a. Séquençage d’exomes chez Integragen sur plateforme Illumina ... 169
b. Traitement bio-informatique des données brutes chez Integragen ... 171
c. Séquençage d’exomes au CNG (Centre National de Génotypage) ... 175
5. Etude bio-informatique des variants identifiés par IVA ... 175
a. Le filtre « Biological Context » (contexte biologique) ... 177
b. Le filtre «Common Variants » (variations communes) ... 177
c. Le filtre « Confidence » (confiance)... 177
d. Le filtre « Genetic Analysis » (analyse génétique) ... 179
e. Le filtre « Predicted Deleterious » (prédit délétère) ... 179
6. Séquençage de Sanger ... 183
a. Méthode de séquençage de Sanger ... 183
b. Validation des variants par séquençage direct ... 185
c. Mise en évidence des pertes d’hétérozygotie (Loss of Function/ LOH) ... 185
7. Analyse de transcrits ... 185
8. Modélisation moléculaire et simulations dynamiques de PDGFRA ... 187
a. Modélisation des domaines extracellulaire D-like des protéines PDGFRα native et mutées 187 b. Simulations moléculaires dynamiques (MD) ... 187
c. Analyse des données de simulations ... 187
9. Essais fonctionnels des variants de PDGFRA ... 189
a. Réactifs et plasmides ... 189
b. Phosphorylation de PDGFRA ... 189
c. Essais luciférase ... 189
d. Incorporation de la thymidine ... 191
10. Analyse génomique à plusieurs niveaux ... 191
a. Hybridation génomique comparatice (CGH) ... 191
1
LISTE DES ILLUSTRATIONS
Figure 1 : Equilibre entre l’action des proto-oncogènes et des gènes suppresseurs de tumeurs Figure 2 : Différents types d’altérations affectant les gènes dans certains cancers
Figure 3 : Evolution des tumeurs par plusieurs étapes génétiques et morphologiques Figure 4 : Les voies de signalisation et les processus cellulaires impliqués dans les cellules cancéreuses
Figure 5 : Hypothèse des deux hits de Knudson
Figure 6 : Faisabilité de l’identification de variants génétiques en fonction de la fréquence allélique et de la force de l’effet génétique
Figure 7 : Les tissus mésenchymateux et leurs dérivés Figure 8 : Classification des sarcomes
Figure 9 : Taux d’incidence des sarcomes en fonction de l’âge Figure 10 : Principales fusions observées dans les sarcomes
Figure 11 : Les réseaux impliqués dans la thérapie ciblée des sarcomes Figure 12 : Fonction de IGF-1 et de IGFBP-3
Figure 13 : Locus CDKN2A et Voies de signalisation incluant RP, p53 et CDKN2A
Figure 14 : Locus CDKN2A et effet de la mutation mise en évidence dans la famille 7389
Figure 15 : Structure du récepteur PDGFRA, principaux sites de mutations dans le GIST et variants identifiés chez nos patients
Figure 16 : Position des variants de PDGFRA détectés
Figure 17 : Caractérisation fonctionnelle des variants de PDGFRA Figure 18 : Effets des variants de PDGFRA sur l’activité luciférase Figure 19 : Expression de PDGFRA mutées dans les cellules transfectées
Figure 20 : Effet des variants de PDGFRA sur la prolifération des cellules en absence de facteur de croissance
Figure 21 : Effet des variants de PDGFRA sur la prolifération des cellules en présence de facteur de croissance
Figure 22 : Prolifération des cellules en fonction du type de ligand
Figure 23 : Profils CGH des 7 sarcomes porteurs de mutations CDKN2A (aCGH)
Figure 24 : Schématisation des voies de signalisation de RB et p53 intégrant CDKN2A et PDGFRA Figure 25 : Les différentes couches de la peau
Figure 26 : La pigmentation de la peau Figure 27 : Développement du mélanocyte
Figure 28 : Migration des cellules de la crête neurale
Figure 29 : Voies de signalisation et enzymes impliquées dans la mélanogénèse et la prolifération du mélanocyte
Figure 30 : Mélanine et exposition aux rayonnements ultraviolets Figure 31 : La pigmentation cutanée
Figure 32 : Spectre morphologique des néoplasmes mélanocytiques
Figure 33 : Evolution de l’incidence du mélanome cutané selon le sexe de 1980 à 2012 Figure 34 : Phototypes cutanés selon la classification de Fitzpatrick
Figure 35 : Gènes à forte, moyenne ou faible pénétrance et leurs localisations chromosomiques Figure 36 : Prévalence des mutations dans les gènes à haut risque de susceptibilité au mélanome Figure 37 : Fonctions biologiques et voies de signalisation des gènes de susceptibilité au mélanome Figure 38 : Les mutations somatiques des gènes impliqués dans les voies de signalisation MAPK et PI3K/AKT
Figure 39 : Classification des néoplasmes mélanocytiques Figure 40 : Classification TNM du mélanome cutané Figure 41 : Mutations de novo
Figure 43 : Série de filtres appliqués à l’aide du logiciel Ingenuity Variant Analysis
Figure 44 : Résumé de Guidelines pour la sélection de variants impliqués dans les maladies humaines ANNEXES :
Figure 45 : Les grandes étapes d’un séquençage d’exomes réalisé chez Integragen Figure 46 : Procédé de capture d’exons réalisé grâce au système SureSelect par méthode d’enrichissement en solution
Figure 47 : Cycle de séquençage par le séquenceur HiSeq 2000 d’Illumina
LISTE DES TABLEAUX
Tableau 1 : Variants constitutionnels identifiés par WES concomitant avec une altération tumorale observée en CGH pour six cas de sarcomes
Tableau 2 : Mutations fréquentes et leur rôle durant la progression d’un mélanome Tableau 3 : série 1 des mélanomes pédiatriques
Tableau 4 : série 2 des mélanomes pédiatriques Tableau 5 : série 3 des mélanomes pédiatriques
Tableau 6 : Variants identifiés par séquençage d’exomes de la première série de mélanomes pédiatriques
Figure 1 : Equilibre entre l’action des proto-oncogènes et des gènes suppresseurs de tumeurs.a A l’état normal, les deux copies du proto-oncogène et les deux copies du gène suppresseur de tumeur agissent de manière équilibrée pour contrôler le cycle cellulaire. Une mutation sur une copie du proto-oncogène est suffisante pour entrainer une prolifération cellulaire excessive. Une mutation sur chaque copie du gène suppresseur de tumeur est nécessaire pour entrainer une prolifération cellulaire excessive.
Prolife ration
cellulaire
normale
Prolife ration
cellulaire
excessive
Prolife ratio
n cellulaire
excessive
5
I. INTRODUCTION
1. La cancérogénèse moléculaire
Les cancers humains sont des processus complexes multifactoriels qui impliquent des dysfonctionnements au niveau de trois grands types de gènes: les proto-oncogènes, les gènes suppresseurs de tumeurs (GST), et les gènes du maintien de l’intégrité génomique, les « care-takers »
1, 2
. Les proto-oncogènes ont été découverts dans les années 1980 par les recherches menées sur les retro-virus cancérigènes. Dans les définitions initiales, les oncogènes étaient des gènes capables de transformer une cellule normale en une cellule maligne, lorsqu’une copie mutée d’un oncogène activé était introduite dans une cellule normale. En sens inverse, un gène était dit « suppresseur de tumeur » (GST) lorsqu’une copie fonctionnelle de ce gène introduite dans une cellule tumorale, permettait la réversion vers un phénotype normal. La 3è catégorie de gènes a été conceptualisée lors de la découverte des premiers gènes de prédisposition aux cancers du côlon, codant pour les protéines de réparation des mésappariements de base de l’ADN, les gènes MSH2, MLH1 et MSH6 3. En effet, la perte de fonction de ces gènes ne répondait pas au critère fonctionnel des gènes suppresseurs de tumeurs car la réintroduction d’une copie fonctionnelle de ces gènes dans une cellule tumorale ne permettait pas la réversion vers un phénotype normal.
Classiquement, on distingue deux grands types de mutations qui sont impliquées dans la cancérogénèse : les mutations sur les gènes « gatekeeper » qui agissent sur les voies principales de contrôle de la population cellulaire, et les mutations sur les gènes « caretaker », qui sont responsables d’une instabilité génétique. Les gènes « gatekeeper » contrôlent la croissance cellulaire en inhibant la prolifération ou amenant différents types de mort cellulaire (sénescence, apoptose,…). Les gènes « caretaker » sont impliqués dans le maintien de la stabilité du génome, ils réduisent le taux de mutations des gènes « gatekeepers ». Ces gènes sont fréquemment mutés aussi bien dans les tumeurs sporadiques qu’héréditaires. Les altérations de ces gènes concourent au développement tumoral suite à des mécanismes génétiques (changement dans la séquence d’ADN) ou épigénétiques (modification de l’expression des gènes par méthylation de l’ADN, remodelage de la chromatine). A l’état normal, il existe un équilibre entre l’expression des proto-oncogènes et des GST. Des mutations au niveau de ces gènes vont rompre cet équilibre et entrainer une prolifération cellulaire à l’origine du développement tumoral (Figure 1).
Figure 2 : Différents types d’altérations affectant les gènes dans certains cancers. On retrouve, selon les cancers, des substitutions de simple base (SBS, single-base substitutions), des petites insertions et délétions (indels), des amplifications, des délétions homozygotes et/ou des translocations.
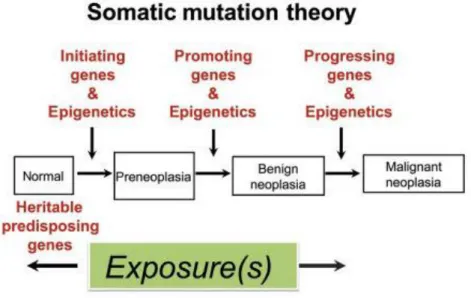
Figure 3 : Evolution des tumeurs par plusieurs étapes génétiques et morphologiques. Les cellules acquièrent des altérations génétiques et épigénétiques, influencées par des facteurs de prédisposition, des expositions environnementales aigues ou chroniques et l’âge.b
7
Les oncogènes, activés ou surexprimés induisent des signaux de prolifération cellulaire excessive ou inappropriée. Les mutations qui affectent ces gènes sont génétiquement « dominantes » ; une mutation activatrice sur un seul allèle est nécessaire pour obtenir un gain de fonction. Les gènes suppresseurs de tumeurs, qui à l’état normal freinent la prolifération cellulaire, n’exercent plus leur contrôle négatif une fois inactivés ; les mutations qui affectent ces gènes sont génétiquement « récessives ». Deux événements génétiques entrainant une perte de fonction sont nécessaires, un sur chaque allèle. Il faut noter que ce même modèle génétique « récessif » est à l’origine de la perte de fonction des « caretakers ».
Dans les tumeurs solides, on observe en moyenne 33 à 66 gènes mutés qui vont donner une protéine dont la fonction est altérée4. Cette moyenne est augmentée (environ 200 mutations) dans le cas de cancers se développant du fait de l’exposition à des mutagènes tels que le mélanome (rayons ultraviolets) ou le cancer du poumon (carcinogènes du tabac). Les mutagènes peuvent aussi être endogènes, stress oxydant, hormones etc ; ils sont alors appelés « facteurs de l’hôte ». Les mutations somatiques identifiées dans les cancers sont principalement des substitutions de simple base (95%), mais on trouve également des petites délétions ou insertions de une à quelques bases, des amplifications, des grandes délétions et des translocations (Figure 2). Pour les substitutions, 90,7 % donnent un changement d’acide aminé alors que 7,6% restent silencieuses et 1,7% touchent des sites d’épissage ou des régions non traduites proches des codons start ou stop.
La transformation cellulaire est un processus réitératif d’expansion clonale, de diversification génétique et de sélection clonale au sein des paysages adaptatifs des écosystèmes tissulaires. Ce processus est complexe avec une possibilité de profils très variables de diversité génétique, d’où l’hétérogénéité génétique des tumeurs5,6. Plusieurs combinaisons différentes d’altérations génétiques peuvent se produire, pour aboutir à des cancers morphologiquement similaires. Ceci contribue à l’hétérogénéité des réponses des patients présentant un même diagnostic clinique. Les tumeurs évoluent donc d’une cellule normale à un statut bénin puis en lésions malignes par l’acquisition d’une série de mutations (Figure 3).
Plus schématiquement et de manière simpliste, une première mutation d’un gène de type « gatekeeper » se produit et confère un avantage sélectif de croissance qui permet le développement d’un petit clone. Puis d’autres mutations se produisent et permettent petit à petit l’expansion clonale, et éventuellement le développement malin de la tumeur qui va envahir la membrane basale sous-jacente et induire des métastases dans les ganglions lymphatiques et les organes à distance.
Figure 4 : Les voies de signalisation et les processus cellulaires impliqués dans les cellules cancéreuses. Tous les gènes drivers connus peuvent être classés parmi douze voies de signalisations (cercle du milieu) qui confèrent un avantage sélectif de croissance (cercle intérieur). Ces douze voies de signalisation peuvent s’intégrer dans trois grands processus cellulaires (cercle extérieur).
9
Ces mutations qui confèrent un avantage sélectif de croissance sont dites mutations « driver ». A côté de ces mutations, on distingue les mutations de type « passenger » qui n’ont pas d’effet sur le processus néoplasique. Ces dernières sont issues de processus aléatoires de mutagénèse, en lien avec des facteurs endogènes, exogènes ou le dysfonctionnement de gènes « caretakers ».
Les mutations à l’origine des cancers sont soit transmises soit induites par des facteurs environnementaux mais elles peuvent également survenir suite à des erreurs lors de la réplication de l’ADN. Il a été montré qu’environ trois mutations se produisent à chaque division de cellules souches et qu’il existe une corrélation entre le nombre de divisions des cellules souches et l’incidence des cancers, indépendamment de l’environnement; ceci démontre que le vieillissement est un facteur de risque du cancer7.
Enfin, il faut noter que des mutations de gènes drivers peuvent se produire au sein de tissus non néoplasiques. Une étude moléculaire récente de lésions d’endométriose, lésions inflammatoires de l’endomètre ayant des caractéristiques de cellules cancéreuses mais non néoplasiques, a montré la présence de mutations « drivers » dans ces lésions, pouvant ne pas évoluer vers une transformation maligne. Ces mutations sont confinées à l’épithélium et n’atteignent pas le stroma, elles doivent donc jouer un rôle clé dans le développement des cellules épithéliales dans les lésions d’endométriose, sans pour autant engendrer une transformation maligne8.
Parmi les altérations génétiques, il existe également des modifications chromosomiques. Beaucoup de tumeurs solides présentent une modification du nombre de chromosomes (aneuploïdie), ainsi que des délétions, des amplifications, des inversions et des translocations. Les translocations aboutissent généralement à la fusion de deux gènes qui soit créé un oncogène, soit permet d’inactiver un GST. Les délétions homozygotes touchent généralement les GST alors que les amplifications se produisent au niveau d’oncogènes. Beaucoup de translocations peuvent également être « passenger », le point de cassure ayant lieu dans des zones dépourvues de gènes ; en effet, les gènes codant des protéines ne représentent qu’environ 1,2% du génome.
Tous les gènes drivers connus peuvent être classés parmi douze voies de signalisation (Figure 4) qui s’intègrent parmi trois grands processus cellulaires : la différenciation cellulaire, la survie cellulaire et la stabilité du génome. Les gènes intervenant dans chacun de ces processus, ou les produits de ces gènes, interagissent entre eux et peuvent amener à des croisements ou des chevauchements de voies de signalisation ; ceci apportant un élément de plus à la complexité du développement tumoral.
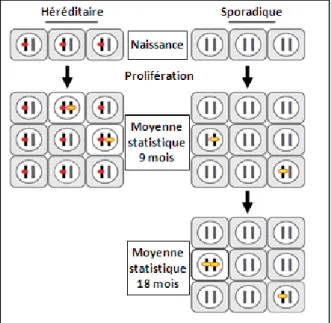
Figure 5 : Hypothèse des deux hits de Knudson. La survenue de deux événements mutationnels (en jaune) dans le cas d’un cancer sporadique nécessite plus de temps que dans les cas héréditaires où une mutation germinale (en rouge) est déjà présente14.
11
Un dernier élément est à considérer dans les processus de cancérogenèse, la plasticité épithélio-mesenchymateuse qui permet la modification des cellules tumorales en divers états phénotypiques lors de leur progression maligne. Cette transition épithélio-mésenchymateuse (EMT) est dépendante de multiples signaux venant du microenvironnement (le stroma). Elle est notamment permise par une régulation de l’expression de gènes clés et notamment une régulation par des mécanismes épigénétiques complexes tels que la modification des histones associées à la chromatine ou la méthylation de l’ADN9.
Au final, parmi toutes les altérations décrites, certaines peuvent être transmises par un gamète parental ou se produire de façon très précoce lors du développement embryonnaire, réduisant ainsi le temps d’accumulation d’anomalies génétiques à l’origine du développement des tumeurs. On parle alors de prédisposition ou de susceptibilité génétique.
2. La prédisposition génétique
La notion de prédisposition est suggérée la première fois, en 1866, lorsqu’un neuroanatomiste et anthropologue français, Paul Broca observe que quinze membres de sa belle-famille ont développé un cancer du sein10. En 1971, Alfred G. Knudson – généticien américain né en 1922 – publie l’hypothèse des « deux hits », c’est-à-dire des deux événements, à l’origine des cancers héréditaires11. Son hypothèse est basée sur des données épidémiologiques qui montrent que les individus atteints de rétinoblastome héréditaire développent leurs tumeurs à un âge précoce12. Knudson propose alors que dans les formes héréditaires de rétinoblastome, une mutation germinale s’est déjà produite et qu’il suffit d’un seul événement supplémentaire dans la cellule somatique appropriée, dans ce cas le rétinoblaste, pour développer une tumeur, à un âge jeune. Dans les cas sporadiques non héréditaires, deux événements mutationnels dans un rétinoblaste sont nécessaires, d’où l’apparition de tumeurs, chez un sujet plus âgé. La nécessité de deux événements plutôt que d’un seul est une des explications de l’observation que les tumeurs mettent plus de temps à apparaître dans les cas somatiques que dans les cas héréditaires (Figure 5). Deux ans plus tard, David Comings proposa que les deux évènements de la cancérogenèse des rétinoblastomes pourraient entraîner la perte de fonction des deux allèles d’un gène dont la fonction normale serait de freiner la prolifération13. Comings insérait cette hypothèse dans le cadre d’une théorie très générale des mécanismes moléculaires de la cancérogenèse conçue comme la conséquence d’une rupture d’équilibre entre deux familles de gènes qu’on appelle aujourd’hui oncogènes et gènes suppresseurs de tumeurs.
13
Les travaux théoriques de Knudson et Comings ont ouvert la voie à la découverte des gènes suppresseurs de tumeurs qui sont effectivement impliqués dans une majorité des syndromes de prédisposition génétique au cancer.
En 1986, RB1, le gène de prédisposition au rétinoblastome et premier gène dit « suppresseur de tumeur » fut identifié, avec dans les cas héréditaires, un allèle muté dans les cellules germinales et le second allèle inactivé de façon somatique15.
On connaît à ce jour plus de cent gènes de prédisposition au cancer10 et on estime que 5 à 10 % des cancers sont d’origine héréditaire16. La plupart des gènes majeurs de prédisposition au cancer ont été identifiés entre 1986 et 2000, par des approches combinant des études de liaisons dans de grandes familles à cas multiples de cancer et du clonage positionnel. Quelques gènes ont été identifiés par des hypothèses de gènes candidats sur des bases fonctionnelles, tels que les gènes de prédisposition au cancer du côlon, MSH2 17 puis MLH1, MSH6 et PMS2. La découverte de ces quatre gènes a posé les bases du concept que des gènes codants des protéines agissant dans une même voie métabolique pouvaient être à l’origine d’un phénotype commun.
Plus récemment, un autre mécanisme que celui des mutations génétiques a été impliqué dans la prédisposition génétique au cancer. Initialement, il a été décrit dans des syndromes de Lynch, des épimutations du gène MLH1, qui correspondent à une hyperméthylation allèle-spécifique du promoteur de ce gène présente dans tous les tissus somatiques, entrainant une diminution d’expression allèle-spécifique. Cette hyperméthylation peut-être dûe à des épimutations primaires, se produisant de novo, responsables de formes sporadiques car effacées lors de la génération suivante. Alternativement, il peut s’agir d’epimutations dites secondaires car résultant d’altérations génétiques, par exemple, d’un SNV dans le promoteur du gène MLH1. Ces dernières sont transmissibles et donc à l’origine de formes familiales de cancers18. Plus récemment, une hyperméthylation du promoteur du gène BRCA1 a été décrite suite à un évènement (non identifié)
de novo post-zygotique chez une jumelle qui a eu une leucémie vers 5 ans puis un cancer de la
thyroide à 22 ans19. Enfin, des hyperméthylations du promoteur du gène SDHC avec perte d’expression, mais sans mutation par séquençage de 130kbp non codants, ont été très fréquemment retrouvées dans des GIST souvent pédiatriques (sans mutation constitutionnelle des gènes SDHx)20.
Les gènes de susceptibilité au cancer peuvent être classés en trois groupes en fonction de la pénétrance des mutations qui s’y produisent (la pénétrance étant définie comme le pourcentage de personnes ayant un génotype donné, qui présentent le phénotype associé à ce génotype). On distingue les gènes à effet fort pour des mutations à haute pénétrance qui confèrent un risque de développer un cancer de plus de 50%, les gènes à effet modéré quand la pénétrance est
Figure 6: Faisabilité de l’identification de variants génétiques en fonction de la fréquence allélique et de la force de l’effet génétique (odds ratio). Les variants identifiés peuvent être à effet faible, modéré, intermédiaire ou fort, et à des fréquences variables, de très rare à commun. Les variants les plus intéressants sont ceux qui se situent entre les deux lignes pointillées en diagonale21.
15
intermédiaire avec un risque de 20 à 50% et les gènes à effet faible quand la pénétrance est faible avec un risque limité21 (Figure 6).
Depuis 2012, la technologie de séquençage d’exome permet d’obtenir des données de manière agnostique, élargissant ainsi considérablement la possibilité d’identifier de nouveaux gènes de prédisposition au cancer. En effet, les technologies actuelles permettent de séquencer les 34 mb de l’exome humain correspondant à 1,2% du génome codant, pour moins de 1 500€22. Cette méthode a permis par exemple, d’identifier le gène CDKN2B23 et le gène BAP124, comme gènes de prédisposition aux cancers du rein à cellules claires. Cependant, la variété des gènes en cause, la difficulté de faire la part entre des variations neutres de l’ADN (un SNP tous les 300 paires de bases, soit un total moyen de 3 millions par génome) et les variations pathogènes, rendent difficile l’analyse des données de séquençage de masse. Pour distinguer les variations pathogènes, on utilise entre autre des bases de données telles que la base ExAC (Exome Aggregation Consortium) en se basant sur la fréquence du variant qui est un indicateur de son potentiel délétère25. ExAC donne l’accès aux données de séquençage d’exomes de 60 706 individus non apparentés issus de diverses études de maladies spécifiques et d’études génétiques de la population. Cette base a également permis de déterminer un système de classement des gènes d’intérêt en fonction d’un score d’intolérance à la perte de fonction (LOF, Loss of fonction); le concept étant que plus les gènes sont intolérants aux mutations « perte de fonction », plus les protéines codées jouent un rôle important dans le maintien de l’homéostasie cellulaire26.
Les premiers résultats de séquençage d’exomes menés dans les familles à cas multiple de mélanome ont montré le plus souvent l’existence d’une très grande variabilité génétique, de type une mutation/un gène/une famille. Ceci expliquant en partie l’échec des stratégies d’identification de nouveaux gènes de prédisposition au cancer menées entre les années 2000 et 2010, par des études de liaison génétique. Une alternative est de travailler sur un grand nombre de familles, dans le cadre de collaborations internationales. De telles approches ont permis d’identifier récemment, les gènes
POT1, ACD et TERF2IP, comme gènes de prédisposition au mélanome27, 28. Dans le cas de cancers très rares, nous avons adopté une stratégie alternative, en utilisant une dimension biologique dans l’analyse de nos données de séquençage d’exomes constitutionnels.
17
II. CONTEXTE ET OBJECTIFS DES RECHERCHES
L’objectif de mon travail de thèse initié fin 2013, était d’explorer le potentiel de découverte de nouveaux gènes de prédisposition au cancer à l’aide de séquençage d’exomes constitutionnels de cas sélectionnés pour une forte suspicion de l’existence d’une prédisposition génétique. En effet, en raison de la présence constitutionnelle d’un 1er évènement génétique, les présentations cliniques sont particulières telles que la survenue d’un cancer à un âge précoce, l’agrégation dans une même branche familiale d’un même type de cancer (ou de différents cancers appartenant à un même syndrome) ou la présence de tumeurs multiples chez un même patient.
Dans cette optique, nous avons choisi de travailler sur 1) une famille à cas multiples de sarcomes afin d’explorer une agrégation familiale d’un même cancer rare en population générale et 2) une cohorte d’enfants atteints de mélanome (sans histoire familiale de mélanome) dans le contexte de cancers du sujet jeune d’apparence sporadique.
Pour la famille à cas multiples de sarcomes, ce type de situation clinique peut être révélateur d’une prédisposition héréditaire. En effet, les sarcomes sont des pathologies rares, leur étiologie est dans la plupart des cas encore mal connue et les agrégations familiales de sarcomes sont trouvées essentiellement dans des familles porteuses de mutations du gène TP53, ce qui n’était pas le cas pour la famille que nous avons sélectionnée. Ainsi, l’étude de cette famille à trois cas de sarcomes était susceptible de nous apporter des réponses sur la sarcomatogenèse.
En ce qui concerne les mélanomes de l’enfant, leur étiologie est différente de celle des mélanomes de l’adulte. L’étude d’une série de mélanomes pédiatriques pouvait donc nous permettre de comprendre la mélanogénèse, au cours du développement.
Afin de mieux comprendre les résultats qui seront présentés dans cette thèse, nous allons dans un premier temps pour chaque projet, présenter la pathologie abordée et l’état des connaissances actuelles, avant d’exposer les résultats obtenus, les discuter et les mettre en perspective. Les populations étudiées et les méthodes utilisées sont présentées en Annexe 3.
Figure 7 : Les tissus mésenchymateux et leurs dérivés29
Le tissu mésenchymateux est à l’origine des tissus dans lesquels se forment les sarcomes : vasculaire, adipeux, musculaire, osseux…
Figure 8: Classification des sarcomesc
Les sarcomes se répartissent en sarcomes des tissus mous (STS) et en sarcomes osseux ; puis en fonction du tissu où ils se développent.
19
III. PARTIE I : CAS DE FAMILLES À CAS MULTIPLES - FAMILLE À CAS
MULTIPLES DE SARCOMES
Dans cette partie seront présentés les principaux résultats de l’article : « Germline
CDKN2A/P16INK4A mutations contribute to genetic determinism of sarcoma, F. Jouenne et al, 2017, Journal of Medical Genetic, on line »30. Pour situer ce travail dans l’état actuel des connaissances, je présenterais la pathologie abordée, le sarcome ; puis je commenterais les résultats obtenus et publiés, ainsi que ceux non publiés. Je finirais par les perspectives envisagées pour ce travail.
1. Le sarcome
A. Définition
Les sarcomes (nom dérivé du grec sarx, la chair et ôma, la tumeur) sont des tumeurs mésenchymateuses malignes développées aux dépens des tissus de soutien de l’organisme : le tissu conjonctif et les tissus qui en dérivent, le tissu vasculaire, le tissu nerveux et le tissu adipeux (Figure 7). C’est un groupe complexe de tumeurs qui comprend plus de cinquante sous-types, chacun distinct génétiquement, histologiquement et pathologiquement.
B. Formes cliniques et histologiques
La classification des sarcomes a beaucoup évolué et aujourd’hui, la référence officielle est la classification internationale, WHO classification31, dont la dernière édition date de 2013. De manière générale, on distingue les tumeurs des tissus mous ou des parties molles et les tumeurs osseuses (Figure 8).
Les sarcomes des tissus mous ou sarcomes des parties molles regroupent plusieurs sous-types qui sont classés en fonction de l’analogie du type de tissu produit par la tumeur et du type de tissu normal correspondant : fibrosarcome, liposarcome (cellules graisseuses), léiomyosarcome (cellules des muscles lisses), synovialosarcome, histiocytome fibreux malin...31. Les trois quarts des sarcomes des parties molles sont histologiquement classés comme sarcome indifférencié pléomorphe, liposarcome, léiomyosarcome, myxofibrosarcome, synovialosarcome ou tumeur des gaines nerveuses périphériques.
Parmi les sarcomes des tissus mous, on distingue également les sarcomes développés à partir des viscères, tels que les tumeurs stromales gastro-intestinales (gastrointestinal stromal tumors ou GIST) qui sont les tumeurs mésenchymateuses les plus fréquentes du tractus gastro-intestinal. Beaucoup
Figure 9 : Taux d’incidence des sarcomes en fonction de l’âge32
(A) Taux par type de sarcomes en fonction de l’âge ; (B) Taux des trois plus importants sous-types de sarcomes osseux en fonction de l’âge ; (C) Taux des sous-types de liposarcomes en fonction de l’âge.
21
de tumeurs anciennement décrites comme tumeurs des muscles lisses gastriques et des gaines nerveuses sont en fait des GIST.
Les sarcomes osseux sont des tumeurs malignes de l’os. Ils sont caractérisés par une croissance rapide, une haute agressivité locale, un potentiel de récurrence élevé et un risque de métastases allant de 20 à 100 %, en fonction du type histologique et du grade. On distingue trois grades (I, II et III) et, dans ses différents grades, surtout des chondrosarcomes, des ostéosarcomes, des sarcomes indifférenciés et des sarcomes d’Ewing.
C. Incidence et mortalité
Les sarcomes sont des tumeurs malignes rares. Du fait de la difficulté de diagnostic, une estimation précise est difficile. L’incidence annuelle des sarcomes des tissus mous est de 5 pour 100 000 habitants et celui des sarcomes osseux, en Amérique du Nord et en Europe, est de 0,8 pour 100 000 habitants31. Des taux d’incidence légèrement plus élevés ont été rapportés en Argentine et au Brésil (1,5 à 2 pour 100 000 habitants) et en Israël (1,4 pour 100 000 habitants). Les sarcomes affectent différemment les différentes tranches d’âge de population (Figure 9). Ils représentent 20 % des cancers pédiatriques, 10 % des cancers de l’adolescent et du jeune adulte et 1 % des cancers solides de l’adulte. Comme pour les autres tumeurs, un âge précoce de développement de la pathologie est un signe de risque de cancer héréditaire.
Il existe une relation entre d’une part le type de tumeur, les symptômes et la localisation et d’autre part l’âge et le sexe du patient31. Les sarcomes des tissus mous peuvent se localiser dans tout le corps, mais 75 % sont situés au niveau des extrémités (le plus fréquent se situe au niveau de la cuisse). Il existe une légère prédominance chez les hommes. Les incidences varient en fonction de l’âge : les rhabdomyosarcomes surviennent presque exclusivement chez les enfants, les synovialosarcomes principalement chez les jeunes adultes, alors que les sarcomes pléomorphes indifférenciés, les liposarcomes, les léiomyosarcomes et les myxofibrosarcomes dominent chez les personnes âgées. Enfin comme la plupart des tumeurs malignes, les sarcomes des tissus mous deviennent plus fréquents avec l’âge, l’âge médian au diagnostic étant de 65 ans. Environ 10 % des patients présentent des métastases (le plus fréquemment dans les poumons), au moment du diagnostic de la tumeur primaire, et au moins un tiers des patients atteints de sarcomes des parties molles décèdent de maladies liées à la tumeur, pour la plupart de métastases aux poumons.
Pour les sarcomes osseux, le taux d’incidence varie en fonction de l’âge et présente une distribution bimodale. Le premier pic survient durant la deuxième décade de vie alors que le second se manifeste chez les personnes âgées de plus de 60 ans.
23
Le risque de développement d’un sarcome osseux pendant la seconde décade de vie est proche de celui de la population âgée de plus de 60 ans, mais il y a tout de même plus de cas pendant la seconde décade de vie. De manière générale, en cas de métastases, les sarcomes sont de mauvais pronostic.
D. Facteurs de risque
1. Facteurs environnementaux
L’étiologie de la majorité des sarcomes est inconnue, mais quelques facteurs de risque sont connus tels que des cancérogènes, des virus et des radiations ionisantes33, 34.
De nombreuses preuves existent sur l’implication des radiations ionisantes en tant que facteur environnemental : les radioisotopes tels que le strontium-89, l’exposition professionnelle à long terme, les effets de la bombe atomique, les patients traités par radiothérapie… Les sarcomes post-radiation surviennent au moins trois ans après la réalisation de la thérapie. L’incidence de ces sarcomes est reliée à la dose d’exposition, ils surviennent chez des patients qui ont été exposés à une dose de plus de 50 Gy35.
En ce qui concerne les cancérogènes, les chimiothérapies telles que les anthracyclines et les agents alkylants augmentent le risque de sarcomes secondaires à la thérapie d’un cancer. De nombreux produits chimiques peuvent également induire un sarcome (méthylcholanthrène, o-nitrotoluène, benzophénone,…). Une exposition à des pesticides, des insecticides et des herbicides augmente également le risque. Certains milieux professionnels sont plus exposés que d’autres : fermiers, ébénistes, maçons, forgerons et outilleurs.
Enfin, des études sur les animaux ont montré que les virus peuvent être à l’origine de certains types de sarcomes. Par exemple, l’oncogène Src, le 1er identifié a été isolé du virus du sarcome de Rous, en 1982. Le virus de l’herpès humain de type 8 (HHV8 pour Human Herpes Virus), virus sexuellement transmissible, est fortement associé avec un risque augmenté de sarcome de Kaposi, chez les patients avec un syndrome d’immunodéficience humaine (sida).
D’autres facteurs de risque ont été rapportés, mais avec des preuves plus limitées : la présence de lymphœdème, la pratique intensive de sport chez les athlètes de haut niveau, le diabète, l’obésité, les hormones féminines, la présence de fluorure dans l’eau de boisson, la poussée de croissance chez l’adolescent…
25
2. Facteurs héréditaires
Etant donné l’extrême rareté des agrégations familials de sarcomes, le caractère héréditaire de ceux-ci a moins été étudié que pour d’autres cancers tels que le cancer du sein ou le cancer de l’intestin, mais certains éléments sont connus.
En ce qui concerne la génétique des cancers, il faut distinguer comme vu précédemment différents types de gènes mis en cause.
Gènes à pénétrance élevée, à haut risque, à effet fort
Les sarcomes sont fréquents dans des contextes de syndromes familiaux ou d’agrégations familiales de cancers, tels que le syndrome de Li – Fraumeni, le rétinoblastome, la neurofibromatose de type I, le syndrome de Rothmund – Thomson, le syndrome de Gardner et Werner…
Le syndrome de Li – Fraumeni (LFS pour Li – Fraumeni syndrome) est un syndrome rare à mode de transmission autosomique dominant. Il est caractérisé par le développement de tumeurs multiples primaires à un très jeune âge, principalement des sarcomes des parties molles ou des sarcomes osseux, des cancers du sein de la femme jeune, des tumeurs du cerveau et des corticosurrénalomes36, affectant des individus et des membres de leur famille porteurs d’une mutation du gène TP53 dans 50 à 70 % des cas37,38. Selon les critères de Chompret39 proposés et réactualisés en 2009 pour aider les cliniciens à reconnaître un syndrome de Li – Fraumeni, il existe trois situations cliniques qui permettent de poser un diagnostic de LFS : 1) un cas index avec une tumeur appartenant au spectre tumoral du LFS (sarcome des tissus mous, ostéosarcome, tumeur du cerveau, cancer du sein pré-ménopause, carcinome des corticosurrénales, leucémie, cancer du poumon broncho-alvéolaire) avant l’âge de 46 ans ET au moins un apparenté de premier ou deuxième degré avec une tumeur du spectre LFS (excepté le cancer du sein, si le cas index a un cancer du sein) avant l’âge de 56 ans ou avec des tumeurs multiples ; 2) un cas index avec des tumeurs multiples (excepté des tumeurs multiples du sein), dont deux au moins appartiennent au spectre tumoral du LFS et dont la première apparaît avant 46 ans ; 3) patient avec un carcinome des corticosurrénales ou une tumeur du plexus choroïde sans tenir compte de l’histoire familiale.
Dans la littérature, le mélanome n’est pas associé au spectre tumoral du LFS, seules des associations sporadiques et controversées ont été reportées entre le spectre du syndrome de LFS et les cas de mélanomes. Par exemple, un cas de mélanome a été décrit chez une jeune femme de 32 ans qui a présenté un ostéosarcome à 28 ans, porteuse d’une mutation constitutionnelle du gène TP5340.
Le deuxième type de maladie où l’on observe la survenue de sarcomes est le Rétinoblastome « héréditaire ». Comme vu plus haut, c’est une maladie à transmission autosomique dominante rare
27
où il est décrit, dans la forme héréditaire, une mutation constitutionnelle du gène RB1, situé sur le chromosome 13q14. Initialement, un risque élevé de tumeurs primitives secondaires, particulièrement des ostéosarcomes, est reporté chez les adultes survivants à un rétinoblastome héréditaire développé pendant l’enfance41. Dans une revue traitant de la fréquence des tumeurs primitives secondaires à un rétinoblastome, les ostéosarcomes se placent en premier (37,0 %) devant les mélanomes (7,4 %), puis les sarcomes des parties molles (6,9 %), les fibrosarcomes (3,3 %), les chondrosarcomes (3,3 %) et les sarcomes non définis (3,3 %)42. La perte de fonction au niveau somatique du gène RB1 a été documentée dans des rétinoblastomes et des ostéosarcomes15.
Le syndrome de Rothmund – Thomson est associé à un risque augmenté de développement d’ostéosarcomes. Dans ce syndrome, des mutations du gène RECQL4 ont été identifiées chez environ deux tiers des patients43,44. Les individus atteints de neurofibromatose de type 1, mutés au niveau du gène NF1, ont une prédisposition au développement de certains types de tumeurs et notamment des rhabdomyosarcomes dans 1 à 6 % des cas45. Près de la moitié du sous-type de sarcomes connu comme tumeur maligne des gaines des nerfs périphériques (MPNST) survient chez des patients atteints de neurofibromatose de type 1 (NF1), maladie due à une mutation de novo ou héritée d’un des parents du gène NF146.
Gènes avec faible risque de pénétrance, à effet faible
Les études d’association pangénomique (GWAS, Genome Wide Association Study) ont permis d’identifier de nombreux variants fréquents en population générale, dits « communs », prédisposant à une maladie10, 47.
Une étude GWAS réalisée sur les ostéosarcomes a permis d’identifier deux nouveaux locus de susceptibilité48 : GRM4 et un locus situé sur le chromosome 2.
E. Diagnostic
a) Clinique
Le sarcome peut se présenter comme une tuméfaction située au niveau du membre supérieur ou inférieur. La douleur peut également être un signe révélateurd.
Une imagerie de type échographie ou scanner permet de visualiser une tumeur. L’imagerie par résonance magnétique (IRM), ou remnographie, est l’examen - clé dans le sarcome.
Si l’IRM suspecte un sarcome, alors une biopsie sera réalisée, afin de prélever un fragment de tumeur. L’anatomopathologiste analysera le prélèvement de tumeur et fera le diagnostic de sarcome.
Figure 10: Principales fusions observées dans les sarcomes49
Les sarcomes présentant des fusions de gènes spécifiques se divisent en deux groupes: les tumeurs à cellules rondes et les tumeurs à cellules fusiformes. Le diagnostic histopathologique dans chaque groupe est souvent difficile. La détection de translocations chromosomiques spécifiques de ces tumeurs et des transcrits de gènes de fusions associés peut être très utile dans le diagnostic des cas difficiles.
PNET, primitive neuroectodermal tumor
Ewing/PNET
Desmoplastic Small round cell tumor Myxoid liposarcoma Alveolar
rhabdomyosarcoma Extraskeletal myxoid chondrosarcoma Clear cell sarcoma
Synovial sarcoma Dermatofibrosarcoma protuberans Low grade fibromyxoid sarcoma Inflammatory myofibroblastic tumor Infantile fibrosarcoma Alveolar soft part sarcoma
29
Le bilan d’extension de la tumeur sera également réalisé. Celui-ci évaluera l’extension locale et à distance (métastase).
L’IRM est l’examen de référence pour évaluer l’étendue locale du sarcome, mais aussi pour évaluer l’extension de la tumeur à distance ; on réalisera un scanner thoracique et/ou abdominal en fonction de la localisation de la tumeur primitive.
b) Morphologie et immunohistochimie
L’anatomopathologiste, par le biais de colorations standards va évaluer la malignité de la tumeur (densité cellulaire, atypies cellulaires et nucléaires, mitoses, nécrose…) et sa ligne de différenciation, c’est-à-dire le type de tissu élaboré par la tumeur (liposarcome, léiomyosarcome, rhabdomyosarcome…). De plus, grâce à différents marqueurs immunohistochimiques, il pourra également préciser le type de sarcome (h-caldesmone, CD34, CKit, DOG1, …).
c) Biologie moléculaire
Les bases moléculaires du sarcome sont mal connues, du fait de la rareté des sarcomes, et de la difficulté à obtenir des prélèvements pour les analyses génétiques avant chimio ou radiothérapies, mais elles contribuent au diagnostic, à la classification et à l’évaluation pronostique des sarcomes. Alors que l’on distingue environ cinquante sous-types histologiques, il existe environ cent cinquante sous-types moléculaires que l’on divise en deux catégories principales selon des caractéristiques cytogénétiques 50,51 :
la première catégorie correspond aux sarcomes avec des altérations génétiques simples, un caryotype diploïde simple avec quelques réarrangements chromosomiques. Dans ce type de sarcomes, les mécanismes à l’origine de la sarcomatogenèse se répartissent en trois grands groupes : une dérégulation transcriptionnelle due à des protéines de fusion résultant de réarrangements génomiques (Figure 10), des mutations somatiques dans des gènes et des voies de signalisation clés et des anomalies de nombre de copies d’ADN (Figure 11):
o exemple de translocation équilibrée (dans un tiers des sarcomes) : dans le sarcome d’Ewing, on trouve une translocation de type t(11,22), à l’origine de la protéine de fusion EWSR-FLI1 et dans le rhabdomyosarcome alvéolaire (ARMS), des protéines de fusion de type Pax3-FOXO1A ou Pax7-FOXO1A due à des translocations de type t(2,13) et t(1,13). Dans ces deux cas, le domaine de liaison à l’ADN d’un facteur de transcription (Pax3/7 ou FLI1) est fusionné à un domaine transactivateur d’un autre facteur de transcription (FOXO1A ou EWSR) et ceci modifie la transcription ;
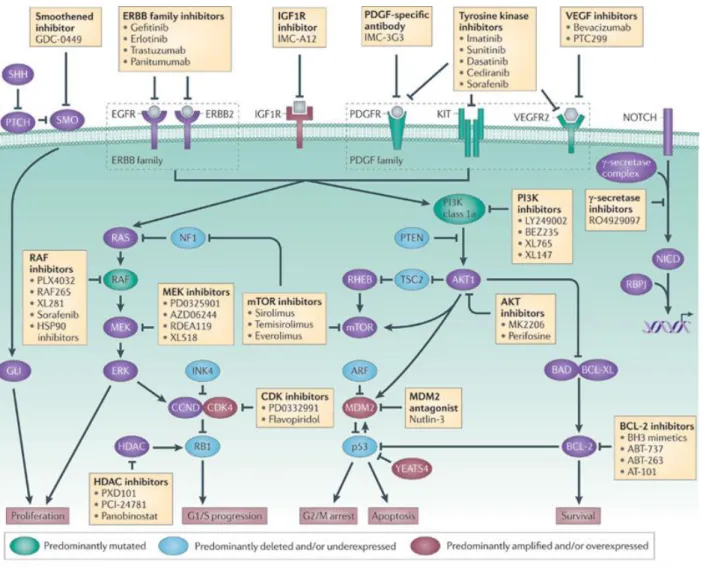
Figure 11: Les réseaux impliqués dans la thérapie ciblée des sarcomes52
Principales cibles des altérations de type mutation ponctuelle, délétion, sous expression, amplification et sur expression observées dans les sarcomes. Les différentes altérations observées, spécifiques des sous-types de sarcomes, impliquent un dysfonctionnement de diverses voies de signalisation : RAS_RAF, PIK3, mTOR, p53, les voies du cycle cellulaire et de la survie, Notch et Hedgehog ; chacune pouvant être ciblées par des thérapies spécifiques. Chaque couleur représente l’altération principale se produisant : vert pour mutation, bleu pour délétion et/ou sous expression, rouge pour amplification ou sur expression.
BAD, BCL-2-associated agonist of cell death; CCND, cyclin D; CDK, cyclin-dependent kinase; EGFR, epidermal growth factor receptor; HDAC, histone deacetylase; HSP90, heat shock protein 90; IGF1R, insulin-like growth factor 1 receptor; NICD, NOTCH intracellular domain; NF1, neurofibromin 1; PDGFR, platelet-derived growth factor receptor; PTCH, Patched; RB1, retinoblastoma 1; RBPJ, recombination signal binding protein for immunoglobulin kJ region; RHEB, Ras homologue enriched in brain; SMO, Smoothened; SSH, slingshot; TSC2, tuberin; VEGFR2, vascular endothelial growth factor receptor 2.
31
o exemple de mutations spécifiques activatrices ou inactivatrices (dans 20% des sarcomes) : c-Kit et PDGFRA dans les tumeurs stromales gastro-intestinales (GIST), SMARCB1/SNF5/INI1 dans les tumeurs rhabdoïdes et les sarcomes épithéloïdes. o exemple d’amplification génique : MDM2 et CDK4 dans les liposarcomes ;
la deuxième catégorie correspond aux tumeurs avec une génétique complexe, c’est-à-dire un caryotype complexe établi par la méthode d’hybridation génomique comparative (CGH,
Comparative Genomic Hybridization) avec des aberrations génomiques multiples
représentatif d’une instabilité génétique. Ce groupe de sarcomes est plutôt observé chez les personnes âgées et inclut des tumeurs telles que l’histiocytome fibreux malin (MFH) ou histiocytofibrome ou le léiomyosarcome où l’on trouve des fréquences relativement élevées de mutations dans les voies de signalisation de p53 et Rb. Il reste encore à identifier des gènes cibles spécifiques à chaque type de sarcome. Les principaux gènes déjà connus comme étant impliqués dans ces sarcomes à génétique complexe sont : PAX3, PAX7, FOXO1, ASPL,
TFE3, FUS, ATF1, EWS, ETV, NTRK3, COL1A1, PDGFB, WT1…
Un grand nombre de sarcomes restent d’étiologie inconnue. L’utilisation de techniques de séquençage à haut débit peut permettre de détecter des anomalies à l’origine du développement tumoral, non détectées par des techniques plus ciblées. A ce jour, seul un séquençage à haut débit d’exomes a été réalisé sur des tissus de sarcomes et a montré l’existence de mutations de gènes majeurs (TP53, PIK3CA, SETD2, AKT1) mais le séquençage d’exomes constitutionnels sur des patients atteints de sarcomes n’avait jamais été réalisé53.
F. Traitement
Le traitement dépendra de la taille de la tumeur, de sa localisation et de son extension à distanced.
La chirurgie, la radiothérapie et la chimiothérapie sont couramment utilisées.
La chirurgie consiste en l’ablation élargie de la tumeur. En effet, une ablation trop limitée expose le patient à une augmentation du risque de rechute locale. Parfois, si la tumeur est étendue ou mal placée et si sa taille ne peut être réduite, le chirurgien peut être amené à réaliser une amputation.
La radiothérapie est souvent réalisée après la chirurgie pour consolider son effet.
La chimiothérapie peut être réalisée avant la chirurgie pour faire diminuer la taille de la tumeur, parfois après la chirurgie, ou parfois être la seule modalité de traitement. En cas de métastases, on utilise des chimiothérapies systémiques, essentiellement la doxorubicine et l’ifosfamide54.
Figure 12: Fonction de IGF-1 et de IGFBP-3
IGF1 se lie à son récepteur IGF-R ce qui déclenche une cascade de réactions en direction du noyau et aboutit à une réponse de survie. En présence d’IGFBP-3, IGF-1 ne se lie pas à son récepteur et la cascade de réaction n’est pas activée. IGFBP-3 a également un rôle, via la liaison à son récepteur IGFBP-3-R, dans l’activation de l’apoptose. En résumé, la présence d’IGFBP-3 va à l’encontre de la survie cellulaire. Ainsi, la diminution de son expression par la protéine de fusion EWSR-FLI1, favorise la survie cellulaire.
33
Du fait de la connaissance des mécanismes moléculaires intervenant dans certains types de sarcomes, des traitements qui agissent de façon spécifique sur les cellules cancéreuses ont pu être développé55. Par exemple, le sarcome d’Ewing est caractérisé par une protéine de fusion, EWSR-FLI1, qui diminue l’expression d’IGFBP-3 (IGF binding protein 3). Cette dernière est une protéine de liaison qui lie sous forme de complexe ternaire et empêche la liaison à son récepteur de IGF1 (Insuline
Growth Factor), peptide-clé de la sarcomatogenèse qui stimule la prolifération cellulaire et inhibe
l’apoptose56 (Figure 12). Il existe de nombreuses preuves d’efficacité des inhibiteurs du récepteur de
l’IGF1 pour bloquer le développement tumoral dans les sarcomes d’Ewing.
Pour le GIST, une mutation ponctuelle est à l’origine de l’activation de récepteurs à activité tyrosine kinases indépendante du ligand : 80 % des cas présentent une mutation sur KIT et 10 % des cas sur
PDGFRA (ces mutations étant mutuellement exclusives). On utilise le mésylate d’imatinib (Glivec®,
Novartis) qui est un inhibiteur de tyrosine kinases et a pour cible, entre autres, KIT et PDGFR. Son efficacité en première ligne dans les GIST a été prouvée par trois essais cliniques randomisés et lui a permis d’obtenir une autorisation de mise sur le marché, en 2001.
De nombreuses recherches et de nombreux essais sont en cours pour produire des thérapeutiques ciblées. En conclusion, l’exploration des mécanismes génétiques à l’origine des sarcomes est un atout majeur pour le développement de ces thérapeutiques.
2. Résumé de l’article
Comme nous l’avons vu précédemment, les sarcomes représentent une pathologie rare dont la pathogénèse est peu connue. Le potentiel du séquençage d’exomes intrafamilial pour identifier des gènes de susceptibilité ayant été démontré, nous avons réalisé un séquençage d’exomes constitutionnel chez deux membres atteints d’une famille à trois cas de sarcomes, où aucune mutation du gène TP53 n’a été mise en évidence. Nous avons exploré les données en appliquant des stratégies classiques de filtres, à l’aide du logiciel d’analyse IVA, et révélé de manière inattendue une mutation, commune aux 3 cas, du gène suppresseur de tumeur CDKN2A. Nous avons également révélé des mutations au niveau des gènes PDGFRA et SKA3.
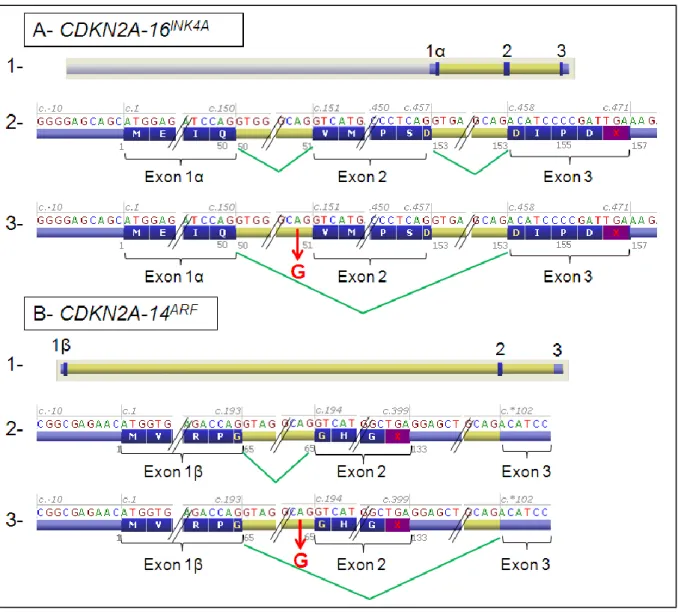
Les mutations du gène CDKN2A sont fréquemment décrites dans les mélanomes « héréditaires »57. Le locus CDKN2A est particulier puisqu’il code deux protéines différentes (Figure 13) : la protéine p16INK4a (Cyclin-Dependent Kinase Inhibitor 2a) qui est impliquée dans la régulation du cycle cellulaire et la protéine p14ARF (Alternative Reading Frame) impliquée dans l’apoptose via p53. Au niveau de l’exon 2, bien qu’il soit partagé par les deux protéines, il y a une phase de lecture qui est différente pour p16INK4a et p14ARF, ainsi on obtient deux protéines totalement différentes avec des rôles