HAL Id: tel-01501306
https://hal.archives-ouvertes.fr/tel-01501306
Submitted on 28 Oct 2020
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Distributed under a Creative Commons Attribution - NonCommercial| 4.0 International License
Fabien Chirot
To cite this version:
Fabien Chirot. Spectrométrie de mobilité ionique et propriétés conformationnelles de systèmes molécu-laires complexes. Chimie théorique et/ou physique. Université Lyon 1 - Claude Bernard, 2017. �tel-01501306�
H
ABILITATION A DIRIGER DES RECHERCHESDélivrée par
L’UNIVERSITE CLAUDE BERNARD LYON 1
soutenue publiquement le 8 février 2017
par
M. Fabien CHIROT
TITRE :
Spectrométrie de mobilité ionique et propriétés
conformationnelles de systèmes moléculaires complexes
JURY : Mme Laurence Charles
M. Philippe Dugourd Mme Marie-Pierre Gaigeot
M. Jérôme Lemoine M. Philippe Maître
Sommaire
Sommaire ... 3
Chapitre I. Physicochimie en phase gazeuse – complexité et structure ... 7
Chapitre II. Mobilité ionique et structure moléculaire ... 13
1. Spectrométrie de mobilité ionique ... 13
2. Quelques bases théoriques ... 15
3. Résolution et forme des spectres de mobilité ... 18
4. Interprétation des résultats et information conformationnelle ... 21
Chapitre III. Etudes structurales par mobilité ionique ... 27
1. Modélisation moléculaire pour la mobilité ionique ... 28
2. Peptides : séquence et conformation ... 30
3. Section efficace – Quelle information structurale ? ... 37
Chapitre IV. Mobilité ionique et spectroscopies d’action – de la combinaison à l’intégration 39 1. Mesures de FRET en spectroscopie d’action et mobilité ionique ... 40
2. Lumière dans le tube de mobilité – développements instrumentaux ... 47
3. Sélectionner, irradier, sonder... 50
Chapitre V. Mobilité en tandem – perspectives ... 59
1. FRET résolu en mobilité ... 59
2. Mobilité ionique et dynamique conformationnelle ... 62
3. Vers des mesures de calorimétrie? ... 67
4. Explorer le paysage conformationnel ... 70
Annexe A. Références ... 73
Annexe B. Informations biographiques ... 78
1. Curriculum vitae ... 78
2. Activités d’enseignement ... 78
3. Responsabilités collectives ... 78
5. Présentations orales dans des congrès depuis 2008 ... 79 6. Liste des publications ... 80 Annexe C. Simulation de profils de mobilité d’isomères en interconversion ... 84
Note sur les références :
Les articles dont je suis co-auteur sont listés dans l’Annexe B.6 et il y est fait référence par un numéro entre crochets dans le texte. Les autres références bibliographiques sont désignées par un numéro en exposant et listées en Annexe A.
Chapitre I.
Physicochimie en phase gazeuse – complexité et
structure
L’ensemble des techniques dérivées de la spectrométrie de masse tire profit de la relative facilité à manipuler des particules chargées à l’aide de champs électromagnétiques, en particulier lorsqu’elles sont sous vide. Depuis ses origines de nombreux progrès technologiques ont contribué à faire de la spectrométrie de masse une méthode analytique de référence, souvent inégalée en termes de précision et de sensibilité. Outre l’amélioration de ses performances métrologiques, une des évolutions les plus importantes de cette technique réside certainement dans la diversification des systèmes qu’elle permet d’étudier. Diversification qui s’est considérablement accélérée depuis la mise au point de méthodes d’ionisation douces telles que la désorption laser assisté par matrice (MALDI)1 et surtout de l’ensemble
des techniques dérivées de l’électro-nébulisation (ESI).2 Ainsi, la gamme des
systèmes accessibles s’étend aujourd’hui des ions atomiques jusqu’aux édifices supramoléculaires de plusieurs millions ou dizaines de millions de Dalton.3,4
Au-delà de son intérêt en tant que méthode analytique, la spectrométrie de masse est un outil de base pour la physicochimie. En premier lieu, elle permet d’isoler des systèmes moléculaires de nature bien définie afin d’étudier leurs propriétés physiques ou chimiques dans un environnement et dans des conditions contrôlés. Il s’agit ensuite de mesurer l’effet sur les ions ainsi préparés d’une perturbation correspondant le plus souvent à une collision avec des molécules neutres, d’autres ions, des électrons, des photons, etc. Un grand nombre de propriétés peut ainsi être étudié, qui inclut la réactivité chimique, les propriétés optiques, électroniques, thermodynamiques. Dans de nombreux cas, la principale observable de l’expérience correspond à un changement de la masse ou de la charge du système. La spectrométrie de masse est donc à la fois utile à définir les conditions de l’expérience et à en déterminer les résultats.
La diversification des objets d’études accessibles s’est naturellement accompagnée de l’application des approches de physicochimie en phase gazeuse à des édifices moléculaires de complexité croissante. Cette diversification n’a donc pas été seulement quantitative mais également qualitative, en ouvrant la voie à l’étude des nouvelles propriétés collectives qui émergent de la complexité des systèmes considérés. Ainsi l’étude de petits agrégats a donné de nombreux exemples de
l’apparition de propriétés à caractère macroscopique avec l’augmentation de leur taille, comme la transition métal-isolant dans les agrégats métalliques.5 Mes travaux
de thèse et ceux que j’ai réalisés au cours de mon stage post-doctoral s’inscrivent dans cette tendance, ayant porté respectivement sur l’apparition d’une transition solide-liquide dans des agrégats de sodium,[29] et sur l’évolution du mode de solvatation d’un électron dans des agrégats d’eau de taille croissante.[28] Ces travaux ont largement reposé sur l’utilisation de la spectrométrie de masse.
Parmi les propriétés d’un système moléculaire, sa structure tridimensionnelle, ou conformation, est l’une de celles qui est le plus intimement liée à sa complexité. C’est autour de l’étude des propriétés conformationnelles d’édifices moléculaires complexes par des techniques dérivées de la spectrométrie de masse que se sont concentrés mes travaux de recherches depuis mon arrivée à l’Université Lyon 1, en 2008. Je me suis en particulier intéressé à une classe de systèmes aux propriétés structurales remarquables : les protéines et les peptides. La structure de ces biomolécules est en premier lieu définie par l’enchaînement des acides aminés qui les composent. Cependant une chaîne d’acides aminés peut se structurer localement pour former différents types d’hélices ou des feuillets-. Ensuite, la conformation dite « native » d’une protéine est définie par l’arrangement de ces sous-structures entre elles. Enfin, un niveau de structuration encore supérieur peut être atteint lorsque plusieurs protéines s’organisent en un complexe supramoléculaire.
L’omniprésence des protéines dans les mécanismes responsables du fonctionnement des organismes vivants, ainsi que leur rôle dans de nombreuses pathologies, justifient à eux seuls l’intérêt d’études structurales. En effet ce sont souvent les propriétés conformationnelles des protéines (capacité à adopter une structure particulière ou au contraire absence de structuration, possibilité de changements de conformation selon l’environnement, etc.) qui leur permettent de remplir une fonction biologique. La capacité de ces systèmes à s’auto-organiser pour former des édifices hiérarchiquement structurés rend toutefois l’étude de leurs propriétés conformationnelles intéressante en soi. D’autant plus qu’une meilleure compréhension des mécanismes qui sous-tendent cette auto-organisation serait profitable non-seulement dans le cadre de la biochimie fondamentale, mais également afin de pouvoir de contrôler ces processus dans un but thérapeutique ou même biotechnologique, par exemple pour la conception de nouveaux matériaux. Les méthodes classiques de biologie structurale ont permis de grandes avancées dans la caractérisation de la structure des protéines et de leurs complexes. En particulier, la cristallographie par mesure de diffraction des rayons X (DRX), et dans une moindre mesure la spectroscopie de résonance magnétique (RMN) ou la microscopie électronique, ont permis d’établir des bases de données structurales pour des dizaines de milliers de protéines avec une résolution atomique. A ces techniques à haute résolution s’ajoutent de nombreuses autres, sensibles à différents aspects de la structure des protéines. Par exemple, la spectroscopie de dichroïsme circulaire permet de quantifier le degré de la structuration secondaire (hélices, feuillets) d’une
protéine, tandis que des mesures de diffraction de rayons X ou de neutrons aux petits angles donnent une estimation du volume global des systèmes étudiés. Ces alternatives aux techniques haute-résolution apportent une information complémentaire tout en ne souffrant pas des mêmes limitations (nécessité d’avoir un cristal pour la DRX, limite en taille pour la RMN liquide, problèmes de purification, etc.). Dans le même esprit, l’utilisation de techniques de phase gazeuse basées sur la spectrométrie de masse commence à émerger dans le cadre de la biologie structurale. Le principal avantage de la spectrométrie de masse dans ce contexte consiste à pouvoir isoler et identifier les différentes espèces qui peuvent co-exister dans un échantillon et dont les contributions individuelles seraient difficiles à extraire d’une mesure en phase condensée. De plus, malgré le développement de méthodes puissantes permettant de visualiser et d’étudier une molécule unique,6 la
spectrométrie de masse reste la méthode la plus précise pour mesurer la composition d’un édifice moléculaire.
S’il paraît évident que les différents niveaux d’organisation d’une protéine ne peuvent être complètement appréhendés par la seule spectrométrie de masse, l’exploitation de celle-ci peut aller bien au-delà de la détermination de la composition des systèmes étudiés. D’une part, la spectrométrie de masse est assez largement utilisée à des fins de séquençage dans le cadre de la protéomique.7 Il s’agit alors de reconstruire
l’enchaînement des acides aminés qui composent une protéine à partir de fragments, obtenus en général après digestion par une enzyme puis fragmentation induite par collisions. Le même principe peut être utilisé pour déterminer la nature et l’organisation interne de protéines ou de complexes non-covalents formés dans le milieu natif. Ce type d’analyses repose sur l’utilisation de réactifs spécifiques (cross-linkers) qui créent de nouvelles liaisons covalentes entre des points se trouvant en contact en solution.8 L’analyse des fragments obtenus permet ensuite de localiser les
régions liées par un cross-linker, et donc en principe d’identifier des points de contact intra- ou inter-moléculaires. Une autre méthode qui rend la mesure de masse sensible à la conformation des protéines en solution consiste à mesurer le taux d’échange proton-deutérium pour une protéine dans l’eau lourde.9 En renseignant
sur la proportion de la surface de la protéine qui est accessible au solvant, ce type de mesure permet de sonder le degré de repliement de celle-ci. Enfin, une spectrométrie de masse dite « native » s’est récemment développée, qui tire parti du fait que les méthodes d’ionisation douces que sont le MALDI et surtout l’ESI permettent de préserver une partie des interactions non-covalentes qui assurent la cohésion des complexes biomoléculaires. La composition et la diversité des édifices moléculaires présents dans un échantillon peuvent ainsi être déterminées assez directement. La gamme de taille accessible par ce type de méthodes dépasse le million de Dalton, ce qui s’approche de la masse d’un virus. 3,4
Les approches décrites dans le paragraphe précédent mettent à profit la capacité de la spectrométrie de masse à sonder la composition et la nature des protéines ou des complexes présents dans un échantillon liquide, et donc potentiellement proche du
milieu biologique. Une démarche alternative consiste à exploiter la possibilité offerte par cette technique d’isoler des systèmes moléculaires de nature et de composition bien définies pour en étudier ensuite les propriétés en utilisant les outils de la physicochimie en phase gazeuse. Parmi ces outils, les spectroscopies d’action dans l’infrarouge permettent d’obtenir une description parfois très fine de l’arrangement moléculaire local. Ainsi des études en spectroscopie de déplétion par double résonance IR/UV ont été menées sur la structure et la dynamique de petits peptides isolés ou solvatés avec une grande résolution spectrale,10 encore améliorée par
l’utilisation de de pièges à ions refroidis.11 D’autre part des mesures de dissociation
induite par l’absorption de photons multiples (IRMPD) ont été réalisées sur une grande variété de biomolécules. L’intérêt de la spectroscopie infrarouge pour ce type de systèmes réside dans sa sensibilité à la présence de liaisons hydrogène intra- ou inter-moléculaires, en particulier à travers les fréquences de vibrations caractéristiques des liaisons N-H et C=O.12,13 En pratique, il reste cependant difficile
d’étendre le domaine d’application de ces techniques au-delà de systèmes modèles de taille relativement modeste. L’interprétation des mesures se heurte en effet à la fois à la congestion des spectres et à la difficulté de réaliser les calculs de chimie théorique nécessaires.
Peu d’autres techniques permettent de rivaliser avec la spectroscopie infrarouge pour l’étude de la structure de biomolécules en phase gazeuse. Même si plusieurs exemples ont montré que les voies de fragmentation d’un peptide après capture d’électrons pouvaient être corrélées à sa structure,14–16 ce type d’expériences semble difficilement
généralisable. Les spectroscopies dans l’UV ou le visible17 semblent constituer une
voie prometteuse, mais sont encore peu développées. En particulier la possibilité d’estimer des distances inter- ou intra-moléculaires par des mesures de transfert d’énergie par résonance de Förster (FRET) dans un spectromètre de masse a récemment été démontrée, d’une part par le groupe de R. Jockush à travers de mesures de fluorescence,18 et d’autre part par le groupe de P. Dugourd par des
mesures de fragmentation.[9]
La technique de phase gazeuse complémentaire de la spectrométrie de masse qui semble aujourd’hui la mieux établie en termes d’études structurales est la spectrométrie de mobilité ionique (IMS). Son champ d’application s’étend des petites molécules organiques aux édifices biomoléculaires les plus complexes accessibles à la spectrométrie de masse. Cette versatilité compense le relatif manque de détail dans l’information structurale qu’elle permet d’obtenir. En effet, la mesure de la mobilité d’un ion moléculaire dans un gaz sous l’action d’un champ électrique ne renseigne en général que sur la forme géométrique globale de celui-ci et ne peut être reliée que de manière ambigüe à sa structure tridimensionnelle. Des mesures d’IMS ont néanmoins permis de mettre en évidence la coexistence de différentes structures dans de petits agrégats et d’étudier les transitions entre ces structures.19–21 Dans le
domaine des biomolécules, les facteurs intrinsèques de stabilisation de différents types de structure secondaire dans des peptides ont pu être déterminés assez précisément.22–24 Dans le cas de protéines et même d’assemblages de protéines, la
combinaison de mesures d’IMS et de simulations moléculaires ont permis de mettre en évidence la préservation de structures natives en phase gazeuse et ont ouvert la voie à l’utilisation de la combinaison de l’IMS et de la spectrométrie de masse comme un outil à part entière pour la biologie structurale.25
Mes travaux de recherche ont porté en premier lieu sur la mise au point de méthodes s’appuyant sur l’IMS à des fins d’études structurales, à la fois à travers des développements instrumentaux et par la construction d’outils d’interprétation adaptés à la technique. Une ligne directrice de ces travaux a été l’exploration de différentes possibilités pour réduire l’ambigüité structurale inhérente à l’IMS par l’utilisation d’autres techniques complémentaires, en particulier spectroscopiques. Cette approche couplée a récemment évolué vers une démarche plus intégrée, visant à mettre en œuvre IMS, sondes optiques et spectrométrie de masse dans un même dispositif expérimental. J’illustrerai dans la suite le déroulement de ces travaux à travers quelques exemples choisis. Ceux-ci recouvrent des développements instrumentaux et théoriques qui sont le fruit de nombreuses collaborations fructueuses, d’abord avec Philippe Dugourd, avec qui je travaille depuis mon arrivée à Lyon, et également avec Florent Calvo, ainsi que de nombreux autres. Avant de revenir sur mes travaux, je m’arrêterai toutefois sur la technique qui en est le centre : la spectrométrie de mobilité ionique.
Chapitre II.
Mobilité ionique et structure moléculaire
1. Spectrométrie de mobilité ionique
Dans le contexte qui nous intéresse ici, le terme mobilité ionique, désigne la facilité avec laquelle un ion peut se mouvoir dans un gaz tampon sous l’influence d’un champ électrique. La spectrométrie de mobilité ionique est donc le pendant en phase gazeuse des différentes méthodes d’électrophorèse qui consistent à séparer des molécules en fonction de leur vitesse de migration dans un électrolyte. Il s’agit donc en premier lieu une technique séparative.
Il est souvent dit que la spectrométrie par mobilité ionique permet de séparer les ions analysés en fonction de leur conformation ou de leur structure. C’est un abus de langage, car le lien entre mobilité et structure moléculaire n’est pas univoque, comme cela sera explicité dans la suite de ce chapitre. Ceci-dit, deux isomères d’un même ion moléculaire ont de fortes chances de n’avoir pas la même mobilité. En effet, d’un point de vue purement géométrique, l’isomère ayant la structure la plus compacte est a priori plus mobile car il offre une surface moins importante pour les collisions avec le gaz tampon, et est donc moins ralenti.
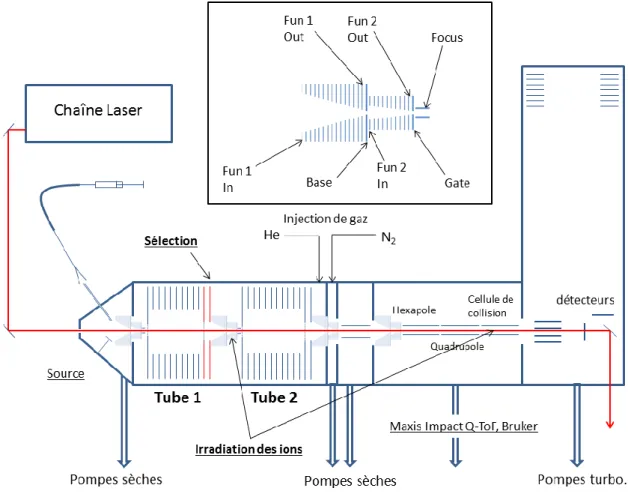
Cette différence s’exprime de manière plus rigoureuse en termes de section efficace. L’aspect équivoque de la notion de compacité permet néanmoins de comprendre l’assez faible spécificité de la mobilité ionique comme grandeur caractéristique pour identifier une espèce moléculaire de manière non ambigüe, en général. Cependant, l’application analytique la plus répandue de la spectrométrie par mobilité ionique est précisément l’identification à des fins de détection de composés volatils, comme des traces d’explosifs ou de drogues. L’avantage de la technique dans ce contexte est sa rapidité, liée au fait que la mesure peut se faire directement dans l’air ambiant où peu d’espèces moléculaires présentent naturellement les mêmes caractéristiques que ces composés. De plus, les spectromètres utilisés peuvent être très compacts (voire portables), et sont en général conçu sur un schéma relativement simple comme présenté sur la Figure II-1, dans lequel la source radioactive utilisée pour la production des ions peut avantageusement être remplacée par une ionisation chimique. En outre, comme la mesure se fait à pression atmosphérique, on peut faire l’économie d’un lourd système de pompage. Le principal enjeu technique pour permettre la robustesse des mesures reste la maîtrise du taux d’humidité, ce qui freine encore le développement de techniques analogues pour l’analyse quantitative de composés volatils dans l’air expiré en vue d’applications médicales.
Dans le cas de systèmes plus complexes en termes de taille moléculaire ou de composition des échantillons, il devient nécessaire de combiner la spectrométrie de mobilité ionique avec une autre technique de mesure complémentaire. C’est en particulier, la combinaison avec la spectrométrie de masse qui a permis d’élargir le champ d’application de l’IMS, aussi bien au niveau analytique, en tant que méthode séparative, que pour son utilisation comme technique d’analyse structurale dans le cadre de la physicochimie fondamentale.
Initialement appelée « chromatographie plasma », la spectrométrie de mobilité ionique s’est d’abord appliquée à l’étude des propriétés de transport d’ions atomiques dans différents gaz. Les caractéristiques des interactions moléculaires en jeu ont ainsi pu être déterminées à partir de modèles théoriques s’appuyant notamment sur les travaux de Nernst sur les électrolytes et de Townsend sur les décharges. C’est dans ce contexte que le cadre théorique de la spectrométrie par mobilité ionique a été formalisé, en particulier dans l’ouvrage de référence de Mason et McDaniel.26 En
combinaison avec la spectrométrie de masse, la technique n’a été appliquée que plus tard, au début des années 1990, à l’étude de la structure de systèmes plus complexes par leur taille ou leur composition. Les travaux précurseurs dans le domaine ont été réalisés dans les groupes de M. Bowers, et de M. Jarrold. L’IMS a ensuite bénéficié de la diversification des objets accessibles à la spectrométrie de masse. L’apparition des sources electrospray2 a ainsi permis d’étudier des biomolécules (peptides, protéines,
fragments d’ADN) jusque-là difficiles à transférer en phase gaz, ouvrant également la voie à de nouvelles applications analytiques. La technique s’est aujourd’hui largement démocratisée depuis la mise sur le marché d’appareils commerciaux couplant spectrométrie de masse et mobilité ionique.
Outre les avancées instrumentales, l’utilisation de la spectrométrie de mobilité ionique pour des études structurales a reposé sur une évolution des outils théoriques pour l’interprétation des résultats. En effet, si la déconvolution des données de mobilité en termes de potentiel d’interaction est relativement directe pour des ions monoatomiques, la dérivation d’une relation avec la structure d’un ion polyatomique
Figure II-1 : Schéma de principe d’un spectromètre par mobilité ionique pour la détection
est plus ardue. La procédure généralement employée consiste à calculer une section efficace à partir d’une structure candidate pour l’ion moléculaire et à comparer cette valeur calculée à une valeur déduite de la mobilité mesurée. Nous reviendrons dans la suite sur les deux difficultés principales liées à la mise en œuvre de cette procédure. Il s’agit d’une part de produire une ou des structures candidates réalistes pour le système étudié. Cela requiert la mise en œuvre d’outils de chimie théorique permettant de localiser les structures les plus probables parmi les nombreuses accessibles à des systèmes aussi flexibles que des protéines. La deuxième difficulté réside dans le calcul d’une section efficace à partir d’une structure donnée. Dans ce domaine l’approche la plus utilisée reste celle mise au point dans le groupe de Jarrold dans les années 1990 qui utilise une intégration Monte-Carlo sur des trajectoires calculées pour des collisions ion-molécule de gaz et s’appuie sur des potentiels d’interaction ajustés sur l’expérience. Nous décrirons plus en détail ces méthodes de calcul à la fin de ce chapitre, après avoir exposé les principales bases théoriques nécessaires pour décrire les phénomènes en jeu dans une expérience de mobilité ainsi que la relation entre mobilité et structure.
2. Quelques bases théoriques
Le contenu de cette section reprend les résultats principaux des différents développements théoriques présentés dans l’ouvrage de référence de Mason et McDaniel26 et synthétisés dans un article de revue paru dans Analytical Chemistry.27
Il s’agit de décrire le mouvement d’un ion dans un gaz sous l’influence d’un champ électrique. Nous ne considèrerons pas ici la possibilité de réaction, en particulier d’échange de charge, entre l’ion et le gaz. Nous resterons de plus dans la limite où le champ électrique est faible. Dans ce cas, la vitesse moyenne de déplacement d’un ion dans le gaz, notée v⃗ D, est simplement proportionnelle au champ électrique 𝐸⃗ :
v⃗ D = 𝐾𝐸⃗ II.1
La relation II.1 définit la mobilité ionique 𝐾 (qui s’exprime en m2.V-1.s-1 dans les
unités du système international). 2.1. Mobilité et diffusion
Un premier résultat très général relie la mobilité définie ci-dessus et le phénomène de diffusion subi par les ions dans le gaz, même en l’absence de champ électrique. La diffusion est décrite par la loi de Fick. Celle-ci relie le flux 𝐽𝑑𝑖𝑓𝑓 d’ions par unité de temps à travers une surface unitaire au gradient local de la densité 𝜌 pour les particules considérées :
𝐽 𝑑𝑖𝑓𝑓 = −𝐷∇⃗⃗ 𝜌 II.2
L’application d’un champ électrique résulte elle aussi en un flux net d’ions dans la direction du champ qui découle de II.1 et qui dépend de 𝜌:
𝐽 𝑐ℎ𝑝. = 𝜌𝐾𝐸⃗ = −𝜌𝐾∇⃗⃗ 𝑉 II.3
où 𝑉 est le potentiel électrique au point considéré.
Les deux relations précédentes étant vérifiées à l’équilibre thermodynamique où le flux net d’ions est nul, on obtient la relation suivante pour la densité d’équilibre 𝜌é𝑞.:
𝐷∇⃗⃗ 𝜌é𝑞.+ 𝜌é𝑞.𝐾∇⃗⃗ 𝑉 = 0 II.4
Or par définition 𝜌é𝑞. doit obéir à une distribution de Boltzmann et dépend donc du
rapport de l’énergie potentielle des ions de charge 𝑞, donnée par 𝑞𝑉 du facteur d’énergie thermique 𝑘B𝑇 :
𝜌é𝑞. = Ae−𝑘𝑞𝑉B𝑇 II.5
où A est une constante, 𝑇 est la température et 𝑘B est la constante de Boltzmann. En différentiant II.5 et en injectant l’expression ainsi obtenue pour ∇⃗⃗ 𝜌é𝑞. dans II.4, on
obtient l’équation suivante, appelée relation d’Einstein, et qui relie la mobilité d’un ion dans un gaz à sa constante de diffusion dans le même gaz :
𝐾 = 𝑞𝐷
𝑘B𝑇 II.6
Cette relation est une illustration du théorème fluctuation-dissipation : le terme dissipatif qui s’oppose au mouvement des ions dans un gaz (lié à leur mobilité) est directement relié aux fluctuations statistiques de position et de vitesse des ions dans ce même gaz (la diffusion) car tous deux ont une origine commune : les collisions entre l’ion et les molécules du gaz. Outre son aspect esthétique, cette relation est d’une grande utilité pratique pour l’interprétation et la déconvolution des spectres expérimentaux (voir paragraphe 3.2).
2.2. L’équation de Mason−Schamp
L’équation II.6 ne permet pas de relier la mobilité d’un ion à sa structure (la constante de diffusion n’étant pas plus explicite à ce sujet). Un tel lien peut être établi en décrivant au niveau microscopique l’effet des collisions avec le gaz sur le mouvement des ions. Dans la limite des conditions habituellement utilisées en spectrométrie de mobilité ionique, il est alors possible de déterminer une relation entre la mobilité d’un ion, ses caractéristiques intrinsèques (masse, charge, géométrie) et les conditions dans lesquelles l’ion évolue (nature, pression et température du gaz, champ électrique). Cette relation est appelée équation de Mason−Schamp :
𝐾 = 3 16√ 2𝜋 𝜇𝑘B𝑇 𝑞 𝑁 1 𝛺 II.7
Ici 𝜇 désigne la masse réduite pour les collisions ion/gaz et 𝑁 est le nombre de molécules de gaz par unité de volume. Ω est la valeur moyenne (sur les orientations et les vitesses initiales accessibles) de la section efficace de transfert d’impulsion pour les collisions de l’ion avec les molécules de gaz. On l’appelle en général, par simplicité, section efficace de collision, souvent abrégé en CCS.
L’équation II.7 exprime de manière explicite l’influence des caractéristiques intrinsèques de l’ion sur sa mobilité. Comme posé de manière intuitive dans le premier paragraphe de ce chapitre, un ion est d’autant plus mobile dans un gaz donné qu’il porte une charge importante. D’autre part la mobilité est inversement proportionnelle à la section efficace 𝛺. Celle-ci dépend bien-entendu des interactions intermoléculaires entre l’ion et le gaz, et donc de la nature chimique de l’ion aussi bien que de celle du gaz. Cependant dans un gaz donné et pour des ions de composition similaire, à plus forte raison pour des isomères, les différences de section efficace sont essentiellement d’origine géométrique et donc liées à des différences de structure. Dans ce contexte, on peut dire en première approximation que 𝛺 est une mesure de la compacité des ions. La mobilité dépend également de la masse de l’ion à travers la masse réduite 𝜇. En pratique cependant lorsqu’on étudie des édifices biomoléculaires, cette dépendance est souvent négligeable. En effet les gaz tampon les plus utilisés sont l’hélium ou le diazote dont la masse moléculaire est bien inférieure à celles des biomolécules et domine donc la masse réduite.
Il est important de garder à l’esprit que l’équation II.7 est établie au prix d’un certain nombre d’approximations et n’est donc valable que dans la limite où les conditions suivantes sont vérifiées :
A. Les interactions entre les ions sont négligeables.
Cette condition est souvent vérifiée en pratique dans le cadre de la spectrométrie de masse.
B. La probabilité de multiples collisions simultanées entre ion et molécules de gaz est faible.
En d’autres termes, le libre parcours moyen des ions dans le gaz doit être grand devant les dimensions géométriques de l’ion. Cela est vérifié pour des édifices moléculaires de masse mesurable en spectrométrie de masse et pour des pressions de gaz inférieures ou égales à la pression atmosphérique.
C. La masse des molécules de gaz est faible devant celle des ions.
Ce qui n’est pas une hypothèse très forte dans le cas de biomolécules dans l’hélium.
D. Le champ électrique est suffisamment faible.
C’est cette condition qui constitue l’hypothèse la plus forte pour obtenir l’équation de Mason-Schamp. Il s’agit de considérer que le mouvement des
ions est dominé par les collisions et consiste donc en une diffusion guidée par le champ électrique. Du point de vue microscopique, cela se traduit par le fait que la quantité de mouvement communiquée à un ion par le champ électrique entre deux collisions est contrebalancée en moyenne par le recul (le transfert de quantité de mouvement) lors d’une collision.
D’un point de vue quantitatif la condition de champ faible s’exprime par une condition sur la valeur du rapport du champ électrique 𝐸 sur la densité 𝑁 du gaz :27 𝐸 𝑁 ≪ 3𝛺2√ 𝑀 𝑀 + 𝑚 𝑘B𝑇 𝑞 II.8
où M et m sont respectivement la masse de l’ion et celle d’une molécule de gaz.
3. Résolution et forme des spectres de mobilité
3.1. Spectre de mobilité ou distribution de temps d’arrivée
Dans une expérience classique de mobilité ionique, les ions se déplacent dans une zone appelée tube de dérive dans laquelle sont maintenus une pression de gaz tampon est constante, ainsi qu’un champ électrique constant. Dans ces conditions, d’après l’équation II.1, le déplacement des ions se fait à vitesse constante.* C’est parce
qu’ils adoptent des vitesses différentes que des ions de conformation ou de charge différentes peuvent être séparés.
Comme la masse et la charge des ions étudiés peuvent souvent être mesurées par spectrométrie de masse et que les conditions expérimentales sont en général raisonnablement contrôlées et mesurables, 𝛺 est la seule inconnue dans l’équation II.7. En combinant les équations II.1 et II.7, il est donc possible de déterminer 𝛺 à partir de la mesure de la vitesse moyenne de déplacement d’un ion dans un champ électrique connu.
En pratique, pour mesurer leur vitesse, on mesure le temps nécessaire aux ions pour parcourir une distance 𝐿 donnée. Dans une expérience classique en tube de dérive les ions sont injectés à un instant donné puis détectés en fonction de leur temps d’arrivée à l’autre extrémité du tube. On peut ainsi établir une distribution de temps d’arrivée, ou spectre de mobilité. Si plusieurs espèces de mobilités différentes sont présentes, plusieurs pics peuvent être distingués dans cette distribution.
Pour l’interprétation des distributions de temps d’arrivée, il est commode d’introduire une variante de l’équation de de Mason-Schamp (II.7) qui peut s’obtenir
* Cette condition n’est pas remplie dans certains types de dispositifs expérimentaux, en particulier
dans les appareils commerciaux de type « travelling wave ».94 C’est une des raisons qui rend difficile la
en combinant (II.1) et (II.7). Elle relie directement le temps de parcours moyen 𝑡D
d’un ion dans un tube de dérive de longueur 𝐿 à sa section efficace Ω : 𝑡D = 16 3 √ 𝜇𝑘B𝑇 2𝜋 𝑁𝐿 𝑞𝐸Ω II.9
3.2. Diffusion et élargissement homogène
Comme dans toute méthode spectroscopique ou spectrométrique, le pouvoir de séparation en spectrométrie par mobilité ionique est lié à la largeur des pics. Pour quantifier ce pouvoir de séparation, on définit la résolution 𝑅 pour un pic de largeur ∆𝑡 centré au temps d’arrivée 𝑡D :
𝑅 = 𝑡D
∆𝑡 II.10
En utilisant la relation d’Einstein (II.6), et en s’appuyant sur la loi de Fick, on peut calculer la contribution de la diffusion à ∆𝑡 :27
∆𝑡𝑑𝑖𝑓𝑓 = 4𝑡D√
𝑘B𝑇ln(2)
𝑞𝑉 II.11
où 𝑉 est la différence de potentiel appliqué sur le tube de dérive.
Cette relation donne la largeur à mi-hauteur de la distribution de temps d’arrivée attendue pour des ions strictement identiques, rigides et injectés exactement au même instant et en un point donné au début du tube de dérive. Comme nous le discuterons dans la suite, d’autres effets influencent en pratique la forme des pics observés. Cependant l’intérêt de ce calcul est de donner la largeur minimale possible pour un pic dans un spectre de mobilité. En combinant (II.10) et (II.11) on peut de plus estimer la résolution maximale qui peut être atteinte dans des conditions expérimentales (𝑉et 𝑇) données et pour des ions de charge 𝑞 donnée :
𝑅𝑚𝑎𝑥 = √ 𝑞𝑉
16𝑘B𝑇ln(2) II.12
3.3. Limites liées à l’instrumentation
Dans le cas idéal où la diffusion constitue la principale contribution à la forme des pics de mobilité, ceux-ci sont Gaussiens avec une largeur donnée par l’équation II.11. En pratique un certain nombre des conditions citées dans le paragraphe précédent ne sont pas réunies. En premier lieu, les ions sont injectés dans le tube de dérive
pendant une durée finie 𝛿. L’influence de la durée d’injection peut être prise en compte en modélisant la durée temporelle initiale du paquet d’ions par une fonction porte. On obtient alors l’expression suivante pour la forme du pic qui résulte de la convolution de cette fonction porte avec la distribution gaussienne dérivant de la loi de Fick : 𝑆(𝑡) =𝐼0 2[𝑒𝑟𝑓 (√ 𝑞𝑉 2𝑘𝐵𝑇 𝑡 − 𝑡𝐷 2𝑡𝐷 ) − 𝑒𝑟𝑓 (√ 𝑞𝑉 2𝑘𝐵𝑇 𝑡 − 𝑡𝐷− 𝛿 2𝑡𝐷 )] II.13
avec erf, la fonction d’erreur I0 et le nombre d’ions entrant dans le tube à 𝑡 = 0. 𝑆(𝑡)
représente le nombre d’ions arrivant au temps 𝑡 à l’extrémité du tube de dérive. Cette modélisation de la durée d’injection des ions permet d’estimer la limite dans laquelle la largeur des pics est dominée par la diffusion, et donc en retour la durée maximale d’injection compatible avec une résolution maximale.
Dans les instruments couplant mobilité ionique et spectrométrie de masse, la détection des ions ne se fait pas directement à l’extrémité du tube de dérive. Ceux-ci doivent être transférés vers un analyseur en masse, souvent via une série plus ou moins longue d’optiques ioniques. Parmi les dispositifs couramment utilisés, les entonnoirs à ions (ion funnels) ou les guides d’ions multipolaires peuvent dégrader la résolution de la mesure en élargissant les pics de mobilité. En effet, une partie des ions peut être piégée, même transitoirement, dans ces dispositifs par les tensions radiofréquences qui leur sont appliquées.
Dans le cas des entonnoirs à ions un autre phénomène peut s’ajouter au piégeage. Les propriétés de focalisation de ces entonnoirs permettent une bonne transmission des ions en sortie d’un tube de mobilité en réduisant l’extension spatiale du nuage d’ions dans la direction perpendiculaire à leur déplacement dans le tube (extension résultant de la diffusion). Or les ions les plus excentrés doivent parcourir un chemin plus long dans l’entonnoir pour être transmis. Cela se traduit par un temps de parcours effectif plus long pour ces ions au moment de la détection.
Ces deux effets indésirables (piégeage et décalage temporel dû à la focalisation spatiale) se traduisent par un élargissement inhomogène des pics de mobilité avec apparition d’une queue du côté des temps les plus longs. Ces phénomènes sont plus difficiles à modéliser que l’élargissement homogène lié à la durée d’injection. En pratique ils peuvent être minimisés en réduisant au maximum (compatible avec la transmission) l’amplitude des tensions radiofréquences utilisées, en utilisant des gradients de potentiel suffisamment importants sur les entonnoirs à ion et en minimisant la pression de gaz résiduel dans les zones de transfert.
4. Interprétation
des
résultats
et
information
conformationnelle
Dans cette section, nous nous intéresserons à l’interprétation de mesures de mobilité ionique en tube de dérive couplée à la spectrométrie de masse. Dans ce cas, pour chaque ion détecté, deux grandeurs sont mesurées directement : son rapport masse-sur-charge (m/z) et son temps d’arrivée (𝑡D) tel que défini plus haut. Les données brutes correspondantes peuvent donc être représentées sur une carte bidimensionnelle. Dans le cadre de l’interprétation structurale des données, cependant, il est souvent plus utile d’extraire une distribution de temps d’arrivée pour chaque valeur de m/z considérée.
4.1. Interprétation directe des spectres
La première information qui peut être extraite directement d’une distribution de temps d’arrivée est liée au nombre de pics présents. Plusieurs pics traduisent la coexistence de plusieurs populations de mobilité différente pour les ions de m/z considérés. L’attribution de ces différents pics à des conformères différents du même ion doit cependant être prise avec précaution.
Présence d’oligomères
D’abord, le rapport m/z considéré peut correspondre à plusieurs espèces de masses et de charges différentes. Par exemple un ion moléculaire chargé deux fois et un dimère de cet ion chargé quatre fois. Il est assez aisé de mettre en évidence ce genre de situations en examinant l’évolution du massif isotopique dans le spectre de masse des ions détectés en fonction du temps d’arrivée (si bien-sûr la résolution en masse est suffisante).
La présence de dimères, ou plus généralement d’oligomères souvent non-spécifiques n’est pas rare en spectrométrie de mobilité ionique. 28 Les conditions de l’analyse sont
en effet relativement douces en raison de la faible vitesse des ions dans le gaz, ce qui peut être un avantage lorsqu’on étudie des complexes non-covalents. Cependant, de nombreuses espèces métastables qui peuvent être conservées lors de l’analyse en mobilité ne résistent pas aux conditions plus dures régnant dans la zone de transfert entre le tube de mobilité et l’analyseur en masse. Les espèces analysées en masse ne sont donc pas nécessairement les mêmes que celles analysées en mobilité. En d’autres termes, si une partie des ions détectés à un m/z donné provient de la fragmentation d’oligomères après l’analyse en mobilité, leur profil de mobilité comporte une contribution de ces oligomères. Cette contribution peut être identifiée aisément à condition qu’une partie des ions parents survive avant l’analyse en masse. Dans le cas contraire, la déconvolution des spectres est plus difficile. Une des possibilités est de tenter de fragmenter d’éventuels oligomères avant l’analyse en mobilité en durcissant les conditions de désolvatation.
Coexistence et interconversion
Lorsque l’ambigüité sur l’assignation des pics à des familles de mobilité différente a été levée, la forme du spectre peut être analysée pour obtenir des renseignements supplémentaires sur les propriétés conformationnelles des ions étudiés. D’abord, la largeur des pics observés peut être comparée à la largeur attendue d’après la résolution de l’instrument. Si le pic est plus large qu’attendu, on peut conclure à la coexistence de plusieurs familles d’ions avec des sections efficaces trop proches pour être résolues. Un exemple courant de ce cas de figure est donné par les spectres de mobilité de protéines.
Lorsque plusieurs familles de structures coexistent, donnant lieu à plusieurs pics de mobilité ou à un seul pic large, une des questions importantes est de déterminer l’origine de ces pics. En particulier, il est important de savoir si ceux-ci reflètent les populations issues du processus de désolvatation et de la composition de la solution de départ ou s’ils sont le résultat d’une interconversion au cours même de la mesure entre les différentes familles présentes. La réponse à cette question n’est pas évidente a priori, et la réalisation d’expériences de mobilité en tandem, ou à des températures différentes21 sont souvent nécessaires. Néanmoins, l’activation des ions avant
l’analyse, ou le changement de la tension d’accélération, conduisant à une modification de la durée de l’analyse peuvent apporter des éléments de réponse dans certains cas (voir Chapitre V.2).
4.2. Mesure de sections efficaces
La détermination de la section efficace à partir de la mesure du temps d’arrivée 𝑡a associé à une famille d’ions donnée s’appuie sur l’application de la formule II.9. Néanmoins le temps 𝑡a n’apparaît pas directement dans cette équation, qui fait intervenir le temps de diffusion dans le tube de mobilité, 𝑡D. Ces deux temps sont
naturellement liés, en effet :
𝑡a = 𝑡D + 𝑡0 II.14
𝑡0 désigne ici le temps de parcours des ions entre la fin du tube de mobilité et le
détecteur. Dans le cas d’une expériences combinant mobilité et spectrométrie de masse, il correspond au temps de transfert vers l’analyseur en masse plus la durée de l’analyse en masse. Le temps 𝑡0 dépend a priori du rapport m/z des ions, de leur
mobilité, ainsi que de la configuration de l’instrument et des paramètres utilisés. Il est donc difficile de prédire sa valeur avec précision. Aussi, la méthode la plus simple et la plus directe pour s’affranchir de cet encombrant paramètre est de réaliser des mesures de temps d’arrivée pour des valeurs différentes du champ électrique 𝐸 appliquée sur le tube de mobilité. Seule la valeur de 𝑡D est modifiée par ces changements. D’après l’équation II.9. 𝑡D varie de manière inversement proportionnelle au champ électrique. Ainsi en traçant le temps d’arrivé mesuré en fonction de l’inverse du champ électrique, on obtient une droite dont l’ordonnée à
l’origine est 𝑡0 et dont la pente permet de déterminer la section efficace Ω en fonction
des conditions expérimentales et de la masse et de la charge de l’ion :
𝑡a(1 𝐸⁄ ) = 𝑡0+16 3 √ 𝜇𝑘B𝑇 2𝜋 𝑁𝐿 𝑞 Ω 1 𝐸 II.15
Cette méthode est en outre relativement précise car la valeur de Ω est déterminée à partir de plusieurs mesures indépendantes. La stabilité des conditions expérimentales, notamment de la pression et de la température du gaz tampon, permet d’obtenir une très bonne linéarité. Il en résulte une faible incertitude sur la détermination de la pente. Par conséquent, la plus grande source d’erreur expérimentale sur la valeur absolue de Ω correspond à l’incertitude sur la mesure absolue de la pression qui est de l’ordre de 2 à 3% avec le dispositif que nous utilisons.
4.3. Calcul de sections efficaces
Au-delà de mesures qualitatives, et de comparaisons entre différents systèmes, la valeur de la section efficace déterminée par mobilité ionique ne peut être interprétée plus avant en termes de structure moléculaire qu’à travers la comparaison avec des structures calculées par des méthodes de chimie théorique. La génération de structures réalistes est un problème en soi et sera discuté plus en détail dans la suite (Cf. Chapitre III.1). Nous nous intéresserons ici au calcul d’une section efficace à partir d’une structure donnée, c’est-à-dire de l’ensemble des coordonnées des atomes composant la molécule, et de la nature de ces atomes.
La section efficace intervenant dans l’expression de la mobilité des ions (voir l’équation II.7) est la section efficace moyenne de transfert d’impulsion pour les collisions entre ion et molécule de gaz, aussi appelée intégrale de collision moyenne. La moyenne porte ici sur l’ensemble de paramètres d’impact, sur la vitesse des molécules de gaz, ainsi que sur toutes les orientations possibles de l’ion lors de la collision. En effet, le champ électrique est trop faible pour orienter les ions dans une direction privilégiée. L’expression générale de la section efficace Ω s’écrit donc de la manière suivante : Ω = 1 8𝜋2∫ d𝜃 2𝜋 0 ∫ d𝜑sin (φ) 2𝜋 0 ∫ d𝛾 2𝜋 0 𝜋 8( 𝜇 𝑘B𝑇) 3 ∫ d𝑣R ∞ 0 e− 𝜇𝑣R 2 2𝑘B𝑇𝑣R5∫ d𝑏2𝑏 (1 ∞ 0 − cos(𝜒(𝜃, 𝜑, 𝛾, 𝑣R, 𝑏))) II.16
Où 𝜃, φ et 𝛾 sont les angles définissant l’orientation de la molécule, 𝑣R est la vitesse
relative de l’ion et de la molécule de gaz, 𝑏 est le paramètre d’impact et 𝜒 est l’angle de diffusion après la collision. Ici le terme exponentiel rend compte de la distribution de vitesse à température 𝑇.
Plusieurs modèles ont été développés pour calculer la valeur de Ω à partir d’une structure donnée et nous ne les décrirons pas ici en détail. L’approximation la plus simple consiste à assimiler la section efficace à la surface moyenne projetée de la molécule. Pour cela, chaque atome de la molécule est considéré comme une sphère dont le rayon est fixé comme paramètre du modèle. Plusieurs jeux de paramètres,29
ainsi que des raffinements du modèle,30 ont été proposés qui permettent de
reproduire relativement fidèlement les résultats expérimentaux, au moins pour des peptides. S’il est clair que la validité de ce type de méthodes est difficile à évaluer, elles présentent néanmoins l’avantage de permettre un calcul très rapide, même pour des édifices moléculaires de très grande taille.31
La méthode de calcul généralement prise comme référence, dite méthode des trajectoires (TM),32 consiste à propager un grand nombre de trajectoires pour des
molécules de gaz entrant en collision avec l’ion d’intérêt afin de déterminer l’angle de déflexion 𝜒 défini dans l’équation II.16. La section efficace est obtenue par intégration en échantillonnant les paramètres d’impact et les vitesses. De plus, la moyenne sur les orientations de la molécule est construite à partir d’un grand nombre de rotations aléatoires. Il s’agit en résumé d’une intégration Monte-Carlo basée sur un calcul de dynamique moléculaire, et donc d’un calcul assez lourd. La particule mobile, la molécule de gaz, se propage dans le potentiel créé par l’ensemble des atomes de l’ion. Le potentiel utilisé est de type Lennard-Jones, et les interactions ion-dipôle induit peuvent être prises en compte à travers la définition de charges ponctuelles sur chacun des atomes. Outre le temps de calcul important, le principal inconvénient de cette méthode est qu’elle repose elle aussi sur une paramétrisation, les deux paramètres de Lennard-Jones devant être définis pour chacun des atomes.
De manière générale, la définition des paramètres constitue la principale faiblesse de chacune des méthodes disponibles.* En effet il est difficile de s’appuyer sur des
résultats expérimentaux car peu de molécules adoptent une structure suffisamment bien définie a priori. De plus il n’est pas évident qu’un jeu de paramètres atomiques établis pour une telle molécule (par exemple C60) soit extrapolable à des molécules de
nature chimique différente (par exemple des peptides). Enfin il ne faut pas oublier la nature du gaz tampon. Si la plupart des expériences en tube de dérive utilisent l’hélium, relativement simple à décrire, la généralisation de l’utilisation de l’azote dans les instruments commerciaux pose de nouveaux problèmes de modélisation.33,34
Il résulte de ces difficultés que la valeur absolue des sections efficaces calculées, quelle que soit la méthode, est à prendre avec précaution. Cette limitation n’est cependant pas rédhibitoire lorsqu’on s’intéresse à des changements de conformation,
* La seule méthode ne souffrant pas a priori de ce problème de paramétrisation semble être celle
développée par A. Shvartsburg qui se base sur le calcul d’angles de déflexion sur une surface d’isodensité électronique.97 Néanmoins les calculs de chimie quantique nécessaires à l’obtention de
ou à la comparaison entre différents systèmes. On s’attend alors à ce que la tendance observée puisse être reproduite, au moins qualitativement, par le modèle.
Chapitre III.
Etudes structurales par mobilité ionique
L’utilisation de méthodes d’ionisation dites douces comme l’electrospray rend possible la préservation d’une partie des interactions intra- et inter-moléculaires faibles, comme les liaisons hydrogène, qui structurent à plus ou moins large échelle les biomolécules et les complexes de biomolécules. Cela a été illustré par l’observation par spectrométrie de masse de complexes non-covalents de protéines.35,36 Des
mesures de mobilité ionique ont de plus été réalisées pour de tels complexes indiquant que leur structure tridimentionnelle semble pouvoir être largement conservée en phase gazeuse.37,38 Cependant, dans le cas de systèmes de taille plus
modeste, comme de petits peptides (>10 acides aminés), de nombreux résultats montrent qu’en l’absence de solvant les interactions électrostatiques dominent.23,39,40[23] La structure observée en phase gazeuse pour ces peptides est
donc essentiellement gouvernée par la répartition des charges et les interactions entre celles-ci, et est donc souvent différente de celle adoptée en solution. De manière assez générale, la structure la plus stable énergétiquement pour une chaine peptidique n’est a priori pas la même en solution et in vacuo. Toutefois, il n’est pas évident que le passage d’une structure à l’autre puisse se faire spontanément lors du processus complexe de désolvatation, ni au cours des différentes étapes précédent l’analyse structurale (qui dépendent du type d’instrument, ainsi que des paramètres expérimentaux utilisés).41 Les propriétés intrinsèques du système entrent également
en ligne de compte, en particulier à travers la possible existence de barrières énergétiques s’opposant à des changements de conformation trop radicaux et qui peuvent conduire à la préservation de structures métastables.
Dans ce contexte, l’extrapolation à la phase condensée des données structurales obtenues en phase gazeuse est pour le moins indirecte.39 Je m’attacherai dans ce
chapitre à montrer qu’il est toutefois possible d’utiliser des mesures en phase gazeuse pour étudier la relation entre la composition d’un édifice biomoléculaire et ses propriétés conformationnelles intrinsèques. En premier lieu, l’analyse directe des spectres de mobilité permet d’évaluer la diversité conformationnelle du système étudié à travers la mise en évidence de la co-exisence ou non de différentes familles de conformères de mobilité différente (voir Chapitre II.4.1). De plus, l’évolution du profil de mobilité d’une espèce donnée en fonction des conditions de préparation de la solution dont elle provient (solvant, pH, température, concentration, etc.) renseigne sur le degré de conservation en phase gaz d’éléments de structure présents en solution.
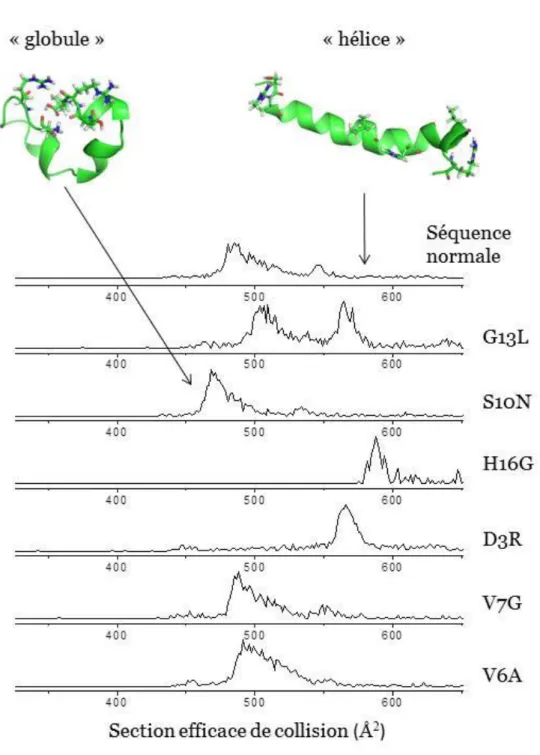
Nous nous sommes intéressés en particulier à l’impact de variations, même ponctuelles, de la nature des acides aminés qui composent une chaîne peptidique sur la flexibilité de cette chaîne, sa propension à former certains types de structures et sur la stabilité de ces structures. Nous avons pour cela adopté une démarche systématique basée sur la comparaison de différents peptides de séquence voisine. Le même type d’approche comparative nous a permis d’examiner l’influence de la topologie d’une chaîne peptidique sur ses propriétés conformationnelles dans le cas de différentes formes isobares d’un peptide antimicrobien.[7] Nous avons également pu mettre en évidence des changements de conformations induits par un processus d’agrégation autour d’une protéine non-structurée, en comparant cette fois les profils de mobilité obtenus en fonction du degré d’agrégation.[20]
L’information conformationnelle la plus directement accessible par des mesures de mobilité ionique concerne la diversité conformationnelle des molécules étudiées, qui peut se refléter dans la coexistence de plusieurs populations de mobilité différentes. Au-delà de cette analyse qualitative, une interprétation des résultats en termes de structure tridimensionnelle nécessite un apport de la modélisation moléculaire. Un des enjeux de telles simulations est de réaliser une exploration efficace du paysage conformationnel complexe de ces systèmes afin de localiser les structures les plus réalistes. Pour ce faire, nous avons réalisé des simulations de dynamique moléculaire basées sur un schéma d’échange de répliques. Nous avons de plus exploré plusieurs approches d’amélioration de l’exploration conformationnelle utilisant les résultats expérimentaux lorsque le schéma précédent n’était pas suffisant.
Dans ce chapitre, nous présenterons d’abord brièvement le principe des simulations de dynamique moléculaire par échange de réplique pour l’interprétation des données d’IMS. Nous discuterons ensuite deux exemples qui illustrent assez bien l’intérêt (et les limites) de la combinaison de l’IMS et de la modélisation moléculaire dans le cadre d’études structurales.
1. Modélisation moléculaire pour la mobilité ionique
*1.1. Exploration conformationnelle
Le but des calculs de dynamique moléculaire que nous avons effectués est en premier lieu d’explorer de la manière la plus exhaustive possible le paysage conformationnel d’une chaîne peptidique afin d’en localiser les régions les plus stables. Le même type de simulations est utilisé dans un second temps pour générer une distribution de populations réaliste à température finie, celle de l’expérience. Il s’agit donc in fine de reproduire au mieux les distributions de temps d’arrivée observées lors des expériences de mobilité ionique.
* Les développements présentés dans cette section sont le fruit d’une étroite collaboration avec Florent
Calvo, qui est notamment l’auteur des codes de dynamique moléculaire ayant permis de réaliser les simulations.
Le modèle choisi, étant donné la taille des systèmes étudiés, est une description des interactions intra- et intermoléculaires par des champs de forces. Nous avons utilisé le champ de forces AMBER99,42 initialement développé pour la phase condensée,
mais assez bien adapté à la phase gazeuse, notamment dans le cas de peptides où les interactions principales sont d’origine électrostatique.
Pour obtenir une exploration efficace, et notamment s’affranchir des barrières énergétiques séparant différent bassins de conformation, nous avons réalisé des simulations en parallèle à différentes températures pour un même système, suivant un protocole développé au départ dans le cadre de simulations Monte-Carlo,43 puis
utilisé en dynamique moléculaire sous le nom de REMD.44 L’intérêt de cette approche
pour l’exploration conformationnelle réside en ce que les trajectoires de dynamique (ou répliques) à haute température permettent de franchir les barrières séparant différentes régions du paysage conformationnel. En revanche, les répliques à plus basse température permettent d’explorer plus finement le « fond » des différents bassins. Pour que cette exploration soit possible sur l’ensemble du paysage accessible, des échanges sont périodiquement tentés entre les structures instantanées de différentes trajectoires adjacentes. Le critère utilisé pour l’acceptation ou non d’un échange est un critère de Métropolis prenant en compte la différence d’énergie entre les structures dans les deux répliques considérées. Ainsi, la probabilité 𝑃é𝑐ℎ.(𝑖, 𝑖 + 1)
d’un échange entre la réplique 𝑖 et la réplique 𝑖 + 1 s’exprime en fonction de la différence entre leurs énergies instantanées 𝐸𝑖 et 𝐸𝑖+1:
𝑃é𝑐ℎ.(𝑖, 𝑖 + 1) = min (1, e(𝐸𝑖+1−𝐸𝑖)(𝛽𝑖+1−𝛽𝑖)) III.1
où 𝛽𝑖 est l’inverse de 𝑘B𝑇𝑖.
Ce schéma permet en principe d’explorer la plupart des minima locaux du paysage conformationnel à l’aide des trajectoires à basse température, les trajectoires à haute température permettant de sauter entre ces minima.
Pour localiser la région de plus basse énergie, un instantané de chaque trajectoire est périodiquement enregistré au cours de la simulation. Une optimisation par une méthode de gradients conjugués est ensuite effectuée pour chacun de ces instantanés, ce qui permet de déterminer les isomères les plus stables. Plusieurs séquences de dynamique et d’optimisation sont successivement menées en initiant une séquence avec la structure la plus stable déterminée lors de la séquence précédente. En pratique, et pour des peptides de 10 à 20 acides aminés, la convergence vers un minimum global putatif est en général atteinte au bout de deux à trois itérations.
1.2. Comparaison avec les résultats expérimentaux
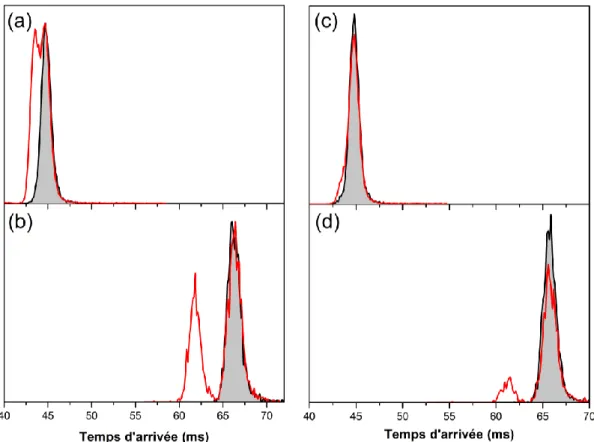
La méthode la plus simple pour comparer les résultats obtenus à l’aide du schéma d’exploration décrit au paragraphe précédent consiste à calculer une section efficace de collision (voir Chapitre II.4.3) pour chacun des isomères de plus basse énergie obtenus lors de l’exploration. L’assignation d’une structure peut ensuite se faire sur
un critère énergétique en cherchant l’isomère de plus basse énergie qui présente le meilleur accord avec la section efficace expérimentale.
Ce protocole simple peut s’avérer efficace lorsqu’une famille de structures se détache de manière marquée en termes de stabilité tout en présentant une section efficace en accord avec la valeur mesurée. La méthode que nous avons utilisée autorise une vision moins statique de la conformation du peptide étudié en ne raisonnant pas seulement en valeur moyenne, mais en cherchant à reproduire de la manière la plus fidèle possible les distributions observées expérimentalement. Il s’agit pour cela d’utiliser différemment les résultats obtenus par les simulations REMD. Pour une dynamique suffisamment longue, l’échantillonnage obtenu pour une trajectoire à température donnée tend en effet vers un ensemble représentatif des populations des différents conformères accessibles. En calculant les sections efficaces de collision pour chacune des structures instantanées, il est donc possible d’obtenir une distribution de sections efficaces. De plus, en utilisant la relation entre la section efficace d’un ion et son temps d’arrivée au détecteur déterminée expérimentalement, on peut simuler la distribution de temps d’arrivée correspondant à la distribution de sections efficaces calculée. Il est même possible de prendre en compte l’effet de la diffusion en convoluant cette distribution par le profil Gaussien émanant des lois de Fick (voir Chapitre II.3.2).
Cette distribution de temps d’arrivée simulée est directement comparable à celle mesurée expérimentalement. Ainsi, en plus d’une simple comparaison entre sections efficaces moyennes mesurées et calculées, il est possible d’évaluer l’impact de la diversité conformationnelle des molécules étudiées sur la largeur des pics dans le spectre de mobilité. Un autre avantage de l’approche présentée ici est qu’en incluant les effets de température elle permet a priori de tenir compte de la contribution d’isomères autres que ceux de plus basse énergie, et plus généralement de l’occurrence de transitions structurales. Nous avons montré dans un article publié en 2012 [18] que de tels effets pouvaient être pris en compte de manière systématique à partir d’outils statistiques, comme la re-sommation d’histogrammes extraits de la simulation REMD.45
2. Peptides : séquence et conformation
*2.1. Approche systématique pour de petits peptides
Nous nous sommes intéressés de manière assez systématique à l’effet conformationnel de la variation d’un acide aminé dans une séquence peptidique, qui ne s’accompagne pas nécessairement d’une modification dans la localisation des charges. Nous avons choisi pour cela une série de poly-alanines de séquence RAAAA-X-AAAAK, ou X est l’un des 20 acides aminés naturels. La présence d’acides aminés
* Les peptides présentés dans cette section ont été étudiés dans le cadre d’une collaboration avec Yury
O. Tsybin, initialement dans le but d’obtenir un jeu de modèles pour étudier la dépendance en conformation de la fragmentation induite par capture d’électrons.
basiques aux deux extrémités du peptide permet ici une connaissance a priori des sites de protonation les plus probables si l’on considère les espèces deux fois chargées positivement de type [M+2H]2+. A part dans le cas où l’acide aminé central est
lui-même suffisamment basique pour entrer en compétition avec l’arginine et la lysine, cela présente l’avantage de simplifier la construction des structures modèles pour les simulations (le champ de forces utilisé impose une localisation définie pour les charges). D’autre part, la localisation des charges permet d’examiner l’effet de la nature de l’acide aminé central sur la conformation du peptide au-delà des effets Coulombiens.
La Figure III-1 reproduit les distributions expérimentales et simulées obtenues pour ces peptides. A l’exception des peptides comportant un acide aminé central basique, pour lesquels la localisation des charges est problématique et que nous omettrons dans la suite, la tendance observée expérimentalement est bien reproduite par les calculs. Cela nous a permis d’examiner plus en détail la distribution des structures calculées et d’évaluer leur diversité conformationnelle.
Dans la plupart des cas on observe une structuration en hélice alpha, plus ou moins étendue selon la nature de l’acide aminé central, à l’exception notable du peptide contenant une proline. Celui-ci, fortement contraint par cet acide aminé particulier adopte une structure plus globulaire. Dans les autres cas, la longueur de l’hélice ne semble pas dépendre de la tendance de chaque acide aminé à se trouver dans des hélices alpha telle qu’elle peut être établie empiriquement en solution.46 En effet les
structures identifiées sont essentiellement affectées par la taille de l’acide aminé central et par la capacité de sa chaîne latérale à solvater les charges présentes aux extrémités du peptide. Ainsi la présence de groupes cabonyl ou carboxyl dans la chaine latérale d’acides aminés tels que Asp ou Glu, favorise l’apparition de motifs de solvatation intramoléculaire des charges qui incluent ces chaînes latérales. D’autre part, les acides aminés les moins encombrants stériquement (Ala, Gly) autorisent un repliement partiel aux extrémités du peptide favorisant la solvatation des charges par les groupements carbonyl du squelette peptidique. Ce repliement est minimisé par la présence d’acides aminés à la chaîne latérale plus massive, comme Trp.
Outre le dégagement de tendances, rendu possible par l’approche systématique adoptée, cette étude a permis de mettre en évidence l’intérêt d’une analyse statistique des distributions obtenues dans les simulations. Par exemple, nous avons pu montrer que la coexistence de différentes familles de structures distinctes dans les simulations pouvait passer complètement inaperçue dans une distribution de sections efficaces.
Figure III-1 : Distributions de sections efficaces mesurées (ligne grise) et simulées (ligne noire) pour
des peptides deux fois protonnés de séquence RA4-X-A4K. La nature de l’acide aminé central est
indiquée à droite des profils correspondants. Les traits pointillés représentent les différents schémas de protonation considérés dans les simulations dans le cas d’un acide aminé central basique.[23]
2.2. Un cas plus complexe : M2TMP
Dans un esprit moins systématique, nous avons utilisé le même type de démarche pour étudier une série de peptide deux fois plus longs que les précédents (25 acides aminés) issue une sous-unité de la protéine M2 du virus de la grippe A. Sous forme d’un tétramère, cette séquence constitue un canal ionique transmembranaire. Certaines mutations ponctuelles ayant été identifiées comme liées à une modification