Comparing phonon dephasing lifetimes in diamond using Transient Coherent Ultrafast Phonon Spectroscopy
Texte intégral
Figure

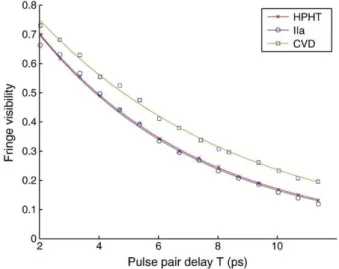
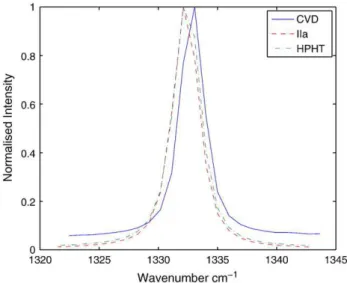
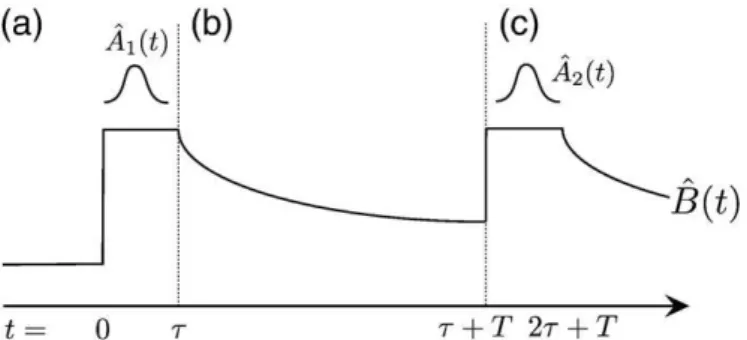
Documents relatifs
he experimental measurement of phonon flux versus propagation angle is achieved by recent extension of the heat-pulse technique known as phonon imaging. /4/ In his method a
for collective excitation self-trapping:first,longitudinal phonons exist with a long lifetime in both phases [as "rotons" at large wavevectors in the superfluid)..
Using this structure factor momentum selection scheme, we can also explain why the 580 cm-l peak of the density of states of the graphite phonons does not show up in the Raman
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des
Moreover, by the use of metal-coated cylindrical or spherical sub-micron hollowed regions on the end of an immersed tapered fibre in a standard near-field scanning optical
The spectra from the debris ejected from both the olive oil and the silicone oil scaifes during the first minute of polishing show traces of oil and diamond (figures 3 & 4)
contrary to their brightness in the optical absorption spectrum, and (ii) strong quantum interference between the first two excitonic reso- nances mediated by nonadiabatic
Polarization of a paramagnet by a fast high intensity magnetic field pulse : spin and phonon relaxation, phonon spectroscopy... POLARIZATION OF A PARAMAGNET BY A FAST
